Phenotypic vs. Target-Based Drug Discovery: A Modern Guide to Strategies, Successes, and AI Integration
This article provides a comprehensive analysis of the two dominant paradigms in pharmaceutical research: phenotypic drug discovery (PDD) and target-based drug discovery (TDD).
Phenotypic vs. Target-Based Drug Discovery: A Modern Guide to Strategies, Successes, and AI Integration
Abstract
This article provides a comprehensive analysis of the two dominant paradigms in pharmaceutical research: phenotypic drug discovery (PDD) and target-based drug discovery (TDD). Aimed at researchers and drug development professionals, it explores the foundational principles of each approach, detailing their methodological workflows and key technological advancements, including the integration of AI and multi-omics data. The content addresses common challenges such as target deconvolution in PDD and efficacy attrition in TDD, offering practical troubleshooting and optimization strategies. Through a critical comparative analysis of success rates, particularly for first-in-class medicines, and an examination of emerging hybrid models, this article serves as a strategic resource for selecting and optimizing drug discovery pipelines for complex diseases.
Defining the Paradigms: The Core Principles of Phenotypic and Target-Based Drug Discovery
What is Target-Based Drug Discovery (TDD)? A History of Rational Design
Target-based drug discovery (TDD) represents a paradigm shift from traditional phenotypic approaches to a rational, hypothesis-driven framework centered on modulating specific molecular targets. This whitepaper delineates the historical emergence, core principles, and methodological workflows of TDD, contextualizing it within the modern drug discovery landscape alongside phenotypic drug discovery (PDD). The transition to TDD was catalyzed by advancements in genomics and molecular biology, enabling high-throughput screening of compounds against isolated proteins implicated in disease pathways. We provide a comprehensive examination of TDD's experimental protocols, key reagents, and strategic advantages while addressing its limitations in translating in vitro efficacy to clinical success. By synthesizing contemporary research and quantitative data, this guide offers drug development professionals a technical resource for navigating target-centric therapeutic development.
Target-based drug discovery (TDD) is a systematic approach to pharmaceutical development that begins with the identification and validation of a specific biological macromolecule—typically a protein or gene—hypothesized to play a critical role in disease pathogenesis. This molecular target serves as the foundational element for all subsequent discovery activities, establishing a causal linkage between target modulation and therapeutic outcome [1]. The TDD paradigm operates on the principle of rational design, wherein drug candidates are deliberately engineered or selected for their ability to interact with a predefined target with high specificity and affinity [2].
The strategic adoption of TDD must be contextualized within the broader dichotomy of drug discovery approaches, particularly in contrast to phenotypic drug discovery (PDD). While PDD identifies compounds based on their observable effects in complex biological systems without presupposition of mechanism, TDD employs a reductionist framework that prioritizes molecular specificity [2] [3]. This target-centric approach gained predominance following the molecular biology revolution and the sequencing of the human genome, which collectively provided an expansive repository of potential therapeutic targets [2]. The fundamental distinction between these approaches has profound implications for screening strategies, lead optimization, and ultimately, clinical translatability.
Historical Emergence of Target-Based Discovery
The evolution of drug discovery methodologies reveals a clear trajectory from serendipitous observation toward rational design. Historically, therapeutic agents were discovered through the empirical screening of natural products or crude extracts in whole organisms—a classical pharmacological approach now categorized as PDD [2] [1]. Seminal examples include morphine from opium poppy and digoxin from foxglove, where therapeutic utility was established long before their molecular mechanisms were understood [1].
The conceptual transition to TDD was catalyzed by several critical scientific advancements. The "one gene, one enzyme" hypothesis proposed by Beadle and Tatum, coupled with the elucidation of DNA's structure, established a mechanistic framework for understanding disease at the molecular level [1]. This foundation enabled pioneering work by researchers such as Gertrude Elion and George Hitchings, who systematically developed purine analogues to intercept specific metabolic pathways, yielding the first antiviral agents and immunosuppressants [1]. Similarly, James Black's rational design of beta-blockers and H₂ receptor antagonists demonstrated the power of targeting specific receptor subtypes to achieve therapeutic selectivity [1].
The completion of the Human Genome Project marked a watershed moment, providing researchers with an unprecedented catalog of potential drug targets and accelerating the pharmaceutical industry's commitment to TDD [2] [4]. Between 1999-2008, however, a surprising observation emerged: a majority of first-in-class drugs were discovered through phenotypic screening rather than target-based approaches [2]. This revelation prompted a re-evaluation of drug discovery strategies, fostering a contemporary perspective that recognizes the complementary strengths of both TDD and PDD within a diversified research portfolio.
Core Principles and Key Characteristics of TDD
The TDD framework is governed by several defining characteristics that distinguish it from phenotypic approaches and establish its rational foundation. Understanding these core principles is essential for effective implementation.
Target-First Hypothesis: TDD initiates with the selection of a specific molecular entity—most commonly proteins such as G-protein-coupled receptors (GPCRs), enzymes, ion channels, or nuclear receptors—that is hypothesized to be causally involved in a disease pathway [1] [4]. This "druggable" target must demonstrate therapeutic relevance, with ideal candidates possessing clear genetic or biochemical evidence linking their activity to disease pathology [4].
Molecular Specificity: A central tenet of TDD is the design of compounds with high selectivity for the intended target over related biological macromolecules [1]. This specificity aims to minimize off-target interactions that could lead to adverse effects, though the therapeutic value of polypharmacology (activity at multiple targets) is increasingly recognized for certain complex disorders [2].
Reductionist Assay Systems: TDD relies predominantly on biochemical or cell-based assays employing purified targets or engineered cell lines with simplified pathophysiology [3]. These systems enable precise measurement of compound-target interactions but may lack the physiological context of native tissue environments.
Define "Established Targets" and "New Targets": The distinction is crucial in TDD. Established targets are those with a well-understood function in normal physiology and disease pathology, supported by extensive scientific literature and often with clinically validated drugs available [1]. In contrast, new targets represent emerging biological understanding with less comprehensive validation, offering potential for first-in-class therapies but carrying greater development risk [1].
The successful prosecution of TDD programs requires rigorous validation of the proposed target's role in disease and its tractability to pharmacological intervention. This process leverages genetic, biochemical, and clinical evidence to establish confidence in the target-disease relationship before committing substantial resources to screening efforts [4].
The TDD Workflow: From Target to Candidate
The implementation of TDD follows a structured, sequential workflow designed to progressively refine compound properties and validate therapeutic hypotheses. The following diagram illustrates the core stages of this process.
Target Identification and Validation
The initial stage involves identifying a biologically relevant molecule with a hypothesized role in disease pathology. Modern approaches leverage genomic analyses (including genome-wide association studies), proteomic profiling, and bioinformatic mining of biological networks to nominate potential targets [4]. Following identification, targets undergo rigorous validation to establish their essential role in disease processes using techniques such as RNA interference, CRISPR-based gene editing, or pharmacological modulation with tool compounds [4] [3]. The emergence of multi-omics integration and machine learning approaches has enhanced the efficiency of this discovery stage [4].
Assay Development and High-Throughput Screening (HTS)
With a validated target, the next phase involves developing robust screening assays capable of interrogating large chemical libraries. TDD typically employs biochemical assays with purified protein targets or cell-based assays employing engineered reporter systems [1]. These assays are optimized for miniaturization and automation to enable high-throughput screening (HTS) of compound libraries ranging from hundreds of thousands to millions of molecules [1]. A critical aspect of this stage is counterscreening, which assesses compound specificity by testing against unrelated targets to eliminate non-selective hits early in the process [1].
Hit to Lead and Lead Optimization
Compounds demonstrating activity in primary screens ("hits") undergo confirmation and preliminary characterization to exclude artifacts or promiscuous inhibitors. Medicinal chemistry efforts then focus on improving the properties of confirmed hits through iterative structure-activity relationship (SAR) studies [1]. Key optimization parameters include:
- Increasing potency against the primary target
- Enhancing selectivity over related targets
- Improving drug-like properties (solubility, metabolic stability, permeability)
- Optimizing pharmacokinetic profiles (absorption, distribution, metabolism, excretion)
This optimization process leverages techniques such as computer-aided drug design, molecular modeling, and structural biology to inform compound design [1]. Contemporary approaches increasingly incorporate fragment-based drug discovery and protein-directed dynamic combinatorial chemistry to explore chemical space more efficiently [1].
Essential Research Reagents and Methodologies
The execution of TDD relies on a specialized toolkit of reagents and methodologies designed to enable precise interrogation of molecular targets. The following table catalogs essential resources for prosecuting target-based campaigns.
Table 1: Key Research Reagent Solutions for Target-Based Drug Discovery
| Reagent/Methodology | Function in TDD | Technical Considerations |
|---|---|---|
| Recombinant Proteins | Purified target proteins for biochemical assays and structural studies | Requires appropriate expression systems (e.g., E. coli, insect, mammalian cells) and functional characterization |
| Engineered Cell Lines | Cellular systems expressing target of interest; may include reporter constructs | Choice of host cell background (e.g., HEK293, CHO) and genetic modification method (transient vs. stable expression) critical |
| Chemical Libraries | Diverse collections of compounds for screening against molecular targets | Library design (diversity, drug-like properties), format (solution, DMSO stocks), and management systems essential |
| Pharmacological Tool Compounds | Reference molecules with established activity at target or related proteins | Used for assay validation, as positive controls, and for understanding structure-activity relationships |
| Target-Specific Assay Kits | Optimized reagents for measuring target activity (e.g., kinase, protease, receptor assays) | Commercial availability, compatibility with HTS formats, and robustness (Z'-factor) influence utility |
| Antibodies | Detection and quantification of target protein expression and modification | Specificity validation (e.g., knockout cell lines) and application compatibility (e.g., Western blot, immunofluorescence) required |
Advanced methodologies that have become integral to modern TDD include DNA-encoded libraries (DELs) for efficient exploration of chemical space, fragment-based screening to identify low molecular weight starting points, and cryo-electron microscopy for structural characterization of challenging targets [1] [5]. The increasing application of artificial intelligence and machine learning further augments these experimental approaches by enabling predictive modeling of compound-target interactions [4] [6].
Comparative Analysis: TDD vs. PDD
The strategic choice between target-based and phenotypic approaches represents a fundamental decision in drug discovery program planning. The following table synthesizes key comparative metrics derived from historical analysis and contemporary research.
Table 2: Quantitative Comparison of TDD and PDD Approaches
| Parameter | Target-Based Discovery (TDD) | Phenotypic Discovery (PDD) |
|---|---|---|
| First-in-Class Medicine Discovery | Lower proportion historically [2] | Higher proportion historically; source of ~50% of first-in-class drugs (1999-2008) [2] |
| Target Space | Limited to previously validated or understood targets [2] | Expands "druggable" space to include unexpected mechanisms and multi-target therapies [2] |
| Screening Throughput | Very high (millions of compounds) [1] | Variable; typically medium to high throughput [3] |
| Mechanism Deconvolution | Inherent to approach | Requires additional target deconvolution efforts; can be resource-intensive [3] |
| Physiological Context | Reductionist; may lack tissue complexity [7] [3] | Higher physiological relevance through use of primary cells, co-cultures, or whole organisms [7] [3] |
| Polypharmacology Assessment | Typically viewed as undesirable (off-target effects) [2] | Can intentionally identify multi-target compounds with potential synergistic effects [2] |
| Technical Success Rate in Primary Screening | Higher probability of technical success [7] | Lower probability due to complex assay systems [7] |
The integration of TDD and PDD represents an emerging paradigm that leverages the strengths of both approaches. This hybrid model may employ phenotypic screening for initial hit identification followed by target-based methods for lead optimization, or conversely, use target-focused assays to characterize compounds discovered in phenotypic screens [3]. The development of more physiologically relevant in vitro systems, including microphysiological systems ("organ-on-a-chip"), 3D organoids, and complex co-cultures, further blurs the distinction between these approaches by enabling target-focused questions to be addressed in more physiological contexts [8] [3].
Target-based drug discovery has established itself as a cornerstone of modern pharmaceutical research, providing a rational, systematic framework for interrogating biological pathways and developing therapeutic agents with defined mechanisms of action. The historical transition to TDD reflected advancements in molecular biology and genomics, enabling unprecedented precision in drug design. Despite challenges in clinical translation, TDD continues to evolve through incorporation of more physiologically relevant model systems, advanced computational methods, and integrative strategies that bridge the divide between target-centric and phenotypic approaches.
The future of TDD will likely be shaped by several convergent trends: the expanding repertoire of "druggable" targets including RNA and protein degradation machinery; the increasing application of artificial intelligence for target validation and compound design; and the growing recognition that polypharmacology may be therapeutically advantageous for complex diseases [2] [5] [4]. For drug development professionals, strategic target selection remains paramount, requiring thoughtful consideration of both biological rationale and practical druggability. As the field advances, the continued refinement of TDD principles—complemented by insights from phenotypic approaches—promises to enhance the efficiency and productivity of therapeutic development.
Phenotypic Drug Discovery (PDD) has re-emerged as a powerful, unbiased strategy for identifying first-in-class therapeutics, marking a significant shift from the reductionist approach of Target-Based Drug Discovery (TDD). This empirical, biology-first approach uses screening methods that do not require prior knowledge of specific molecular targets, instead identifying active molecules based on their effects on cells, tissues, or whole organisms relevant to human disease [9]. The renewed interest in PDD follows a systematic analysis revealing that between 1999 and 2008, phenotypic approaches were responsible for 28 first-in-class small molecule drugs compared to 17 from target-based methods [9] [10]. This surprising finding triggered a major resurgence in PDD adoption, with large pharmaceutical companies like AstraZeneca and Novartis increasing their use of phenotypic screens from less than 10% to an estimated 25-40% of their project portfolios between 2012 and 2022 [9].
Modern PDD should not be confused with historical approaches. Today's PDD leverages very modern tools including high-content imaging, RNA profiling, CRISPR, and advanced computational methods to recreate disease in microplates with higher physiological relevance [10]. This paradigm shift represents a fundamental change in how we conceptualize drug discovery, challenging assumptions about what is druggable and expanding the target space to include unexpected cellular processes and mechanisms of action [2].
PDD vs. TDD: Comparative Analysis and Strategic Implementation
Fundamental Philosophical Differences
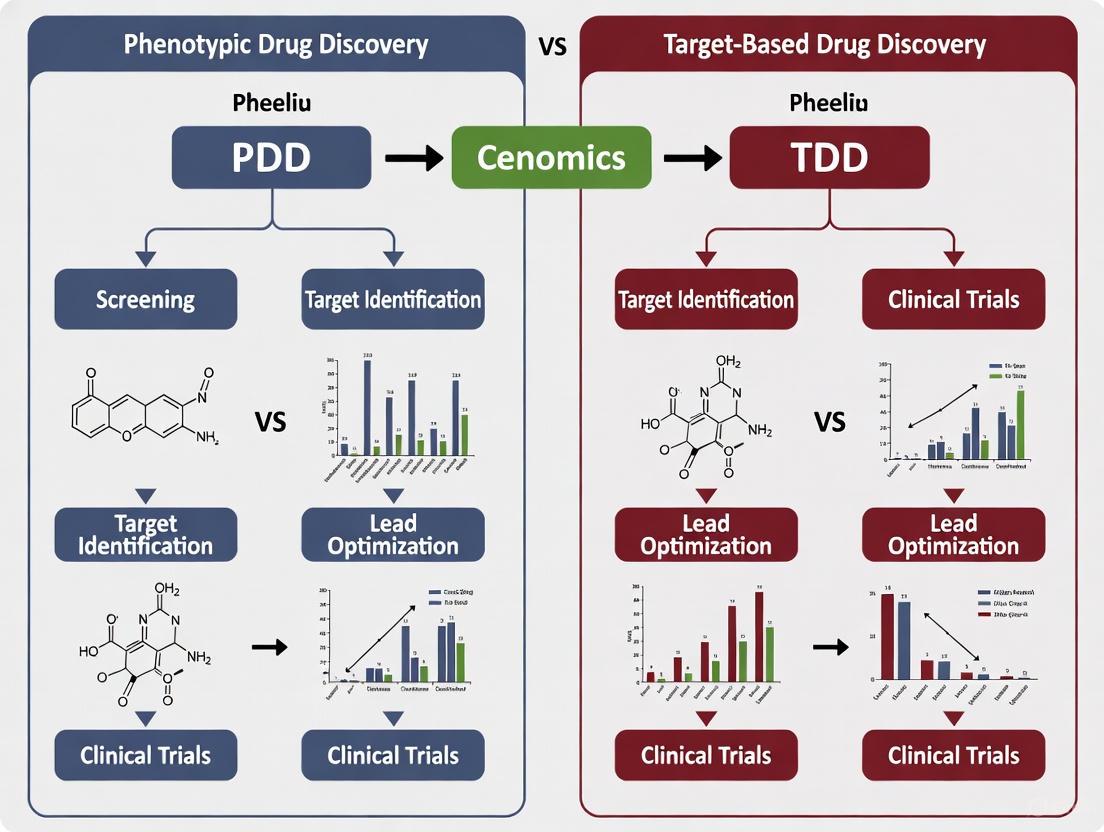
The core distinction between PDD and TDD lies in their starting points and underlying philosophies. TDD begins with a hypothesis about a specific molecular target's role in disease, followed by screening for compounds that modulate this predefined target [11]. In contrast, PDD starts with a disease-relevant biological system and identifies compounds that produce a therapeutic phenotype without requiring target knowledge [9] [11]. This fundamental difference leads to distinct advantages and limitations for each approach (Table 1).
Table 1: Comparison of Phenotypic vs. Target-Based Drug Discovery Approaches
| Parameter | Phenotypic Screening (PDD) | Target-Based Screening (TDD) |
|---|---|---|
| Discovery Approach | Identifies compounds based on functional biological effects in complex systems [11] | Screens for compounds that modulate a predefined molecular target [11] |
| Discovery Bias | Unbiased, allows for novel target identification [11] | Hypothesis-driven, limited to known pathways and targets [11] |
| Mechanism of Action | Often unknown at discovery, requiring later deconvolution [11] | Defined from the outset based on target knowledge [11] |
| Target Space | Broad, includes novel and diverse target types [9] | Narrow, typically limited to enzymes and receptors with known function [9] |
| Success Rate for First-in-Class | Higher proportion of first-in-class medicines [9] [10] | Lower proportion of first-in-class medicines [9] [10] |
| Technological Requirements | Requires high-content imaging, functional genomics, and AI/ML [9] [11] | Relies on structural biology, computational modeling, and enzyme assays [11] |
| Typical Applications | Diseases with complex biology or unknown mechanisms; novel target discovery [12] | Well-validated targets with established biology [12] |
Quantitative Outcomes Assessment
The impact of PDD on drug discovery is demonstrated through quantitative analysis of approved therapies. A comprehensive review showed that from 1999 to 2017, PDD contributed to 58 out of 171 total approved drugs, compared to 44 approvals from TDD and 29 from monoclonal antibody-based therapies [9]. This track record of success, particularly for first-in-class medicines, has solidified PDD's position as a valuable discovery modality in both academia and the pharmaceutical industry [2].
The following diagram illustrates the fundamental differences in workflow between PDD and TDD approaches:
Modern PDD Methodologies: Experimental Frameworks and Workflows
Core Screening Platforms and Model Systems
Modern PDD employs sophisticated biological systems that closely mimic human disease physiology. The selection of appropriate model systems is critical for generating clinically relevant results [10].
In Vitro PDD Platforms have evolved significantly from simple 2D cell cultures to complex, physiologically relevant systems:
- 2D Monolayer Cultures: Traditional cell culture models used for basic functional assays and cytotoxicity screening [11]
- 3D Organoids and Spheroids: More physiologically relevant models that better mimic tissue architecture and function, commonly used in cancer and neurological research [11]
- iPSC-Derived Models: Induced pluripotent stem cells differentiated into specific cell types, enabling patient-specific drug screening and disease modeling [11]
- Patient-Derived Primary Cells: Cells derived directly from patients, offering high clinical relevance for disease modeling [10] [11]
- Organ-on-Chip Models: Systems that recapitulate human physiological processes by merging cell culture with microengineering techniques in microfluidic devices [11]
In Vivo PDD Platforms provide whole-organism context for evaluating therapeutic effects:
- Zebrafish: Small vertebrate model with high genetic similarity to humans, used for neuroactive drug screening and toxicology studies [11]
- Caenorhabditis elegans: Simple, well-characterized organism used in neurodegenerative disease research and longevity studies [11]
- Rodent Models: Gold-standard mammalian models in preclinical research that provide robust data on pharmacodynamics and pharmacokinetics [11]
High-Content Screening and Phenotypic Profiling
High-content screening (HCS) represents arguably the most powerful enhancement to modern PDD, combining automated microscopy with computational image analysis to extract rich morphological data from cells [9]. The Cell Painting assay has emerged as a particularly valuable phenotypic profiling technique, using multiple fluorescent dyes to mark key cellular components and computational analysis to extract thousands of morphological features [13]. This approach enables clustering of cellular phenotypes to help identify potential drug candidates and elucidate mechanisms of action [9].
The experimental workflow for a typical phenotypic screening campaign involves multiple standardized steps:
Essential Research Reagents and Solutions
Successful implementation of PDD requires carefully selected biological tools and reagents. The following table details key research solutions essential for modern phenotypic screening:
Table 2: Essential Research Reagent Solutions for Phenotypic Drug Discovery
| Research Solution | Function in PDD | Application Examples |
|---|---|---|
| Cell Painting Kits | Multiplexed fluorescent dye sets that mark multiple organelles for high-content morphological profiling [13] [14] | Phenotypic profiling, mechanism of action studies, hit triage [13] |
| CRISPR Libraries | Enable genome-wide functional screening to identify genes essential for specific phenotypic responses [10] | Target identification, validation of compound mechanism [10] |
| iPSC Differentiation Kits | Standardized protocols and reagents for generating disease-relevant cell types from induced pluripotent stem cells [11] | Neurological disease modeling, patient-specific screening [11] |
| 3D Culture Matrices | Specialized extracellular matrix materials that support formation and maintenance of organoids and spheroids [11] | Complex disease modeling, tumor biology studies [11] |
| L1000 Assay Reagents | Gene expression profiling technology that measures 978 representative transcripts for low-cost transcriptional profiling [13] | Mechanism of action classification, connectivity mapping [13] |
| High-Content Imaging Reagents | Fluorescent dyes, antibodies, and probes for monitoring cellular processes and morphological changes [9] [11] | Multiparameter phenotypic assessment, live-cell imaging [9] |
Recent Success Stories: PDD-Generated Therapeutics
PDD has contributed to numerous recently approved therapies, particularly for diseases with complex biology or previously undruggable targets. These success stories demonstrate the power of phenotypic approaches to identify first-in-class medicines with novel mechanisms of action.
Table 3: Recently Approved Therapies Identified Through Phenotypic Drug Discovery
| Drug (Brand Name) | Therapeutic Area | Year Approved | Key Mechanism/Target | PDD Approach |
|---|---|---|---|---|
| Risdiplam (Evrysdi) | Spinal Muscular Atrophy | 2020 [9] | SMN2 pre-mRNA splicing modifier [9] [2] | Phenotypic screen for compounds increasing full-length SMN protein [9] |
| Vamorolone (AGAMREE) | Duchenne Muscular Dystrophy | 2023 [9] | Dissociative steroid that modifies downstream receptor activity [9] | Phenotypic profiling to elucidate sub-activities and dissociate efficacy from steroid side effects [9] |
| Lumacaftor/Ivacaftor (ORKAMBI) | Cystic Fibrosis | 2015 [9] | CFTR corrector/potentiator combination [9] [2] | Target-agnostic compound screens using cell lines expressing disease-associated CFTR variants [9] [2] |
| Daclatasvir (Daklinza) | Hepatitis C Virus | 2014/2015 [9] | NS5A replication complex inhibitor [9] [2] | Phenotypic screening using HCV replicon system [2] |
| Perampanel (Fycompa) | Epilepsy | 2012 [9] | AMPA receptor antagonist [9] | Whole-system, multi-parametric modeling in phenotypic assays [9] |
| Lenalidomide (Revlimid) | Multiple Myeloma | 2005 [12] | Cereblon E3 ligase modulator leading to IKZF1/3 degradation [2] [12] | Phenotypic screening of thalidomide analogs for enhanced TNF inhibition [12] |
Case Study: Risdiplam for Spinal Muscular Atrophy
Spinal Muscular Atrophy (SMA) is a rare neuromuscular disease caused by loss-of-function mutations in the SMN1 gene. Phenotypic screens identified small molecules that modulate SMN2 pre-mRNA splicing to increase levels of functional SMN protein [2]. The approved drug, risdiplam, works by engaging two sites at the SMN2 exon 7 and stabilizing the U1 snRNP complex - an unprecedented drug target and mechanism of action [2]. This target would have been unlikely identified through traditional target-based approaches since SMN2 lacked known functional activity relevant to the disease [9].
Case Study: Cystic Fibrosis Modulators
Cystic fibrosis is caused by mutations in the CFTR gene that decrease CFTR function or interrupt intracellular folding and membrane insertion. Target-agnostic compound screens using cell lines expressing wild-type or disease-associated CFTR variants identified both potentiators (such as ivacaftor) that improve CFTR channel gating, and correctors (such as lumacaftor, tezacaftor, and elexacaftor) that enhance CFTR folding and plasma membrane insertion [2]. The combination of elexacaftor, tezacaftor, and ivacaftor was approved in 2019 and addresses 90% of the CF patient population [2].
AI and Machine Learning in PDD
Computational Advances in Phenotypic Analysis
Artificial intelligence and machine learning have dramatically enhanced PDD by enabling automated analysis of complex phenotypic data. ML/AI tools provide significant advantages for PDD through automated analysis of cell image data, extraction of diverse morphological features, and clustering of cellular phenotypes to help identify potential drug candidates [9]. Advanced computational methods can leverage multimodal data, combining chemical structure features with extracted image features to significantly improve the prediction of mechanism of action and bioactivity properties [9].
Recent research demonstrates that combining multiple data modalities dramatically improves bioactivity prediction. One study found that while chemical structures (CS), morphological profiles (MO) from Cell Painting, and gene expression profiles (GE) could individually predict 6-10% of assays with high accuracy (AUROC >0.9), in combination they could predict 21% of assays - a 2 to 3 times improvement over single modalities [13]. At more practical accuracy thresholds (AUROC >0.7), combining modalities increased predictable assays from 37% with chemical structures alone to 64% when integrated with phenotypic data [13].
Emerging AI Platforms and Foundation Models
The field is rapidly evolving with new AI-driven platforms specifically designed for phenotypic discovery:
PhenoModel: A multimodal molecular foundation model using dual-space contrastive learning to connect molecular structures with phenotypic information from cellular morphological profiles [14]. This model outperforms baseline methods in molecular property prediction and active molecule screening based on targets, phenotypes, and ligands [14].
Recursion-Exscientia Integrated Platform: Following their 2024 merger, this integrated platform combines Recursion's extensive phenomics data with Exscientia's generative chemistry capabilities, creating a closed-loop design-make-test-learn cycle powered by automated robotics and AI [15].
Ardigen's phenAID Platform: Dedicated to reducing analysis time and enhancing prediction quality for high-content screening datasets through advanced machine learning algorithms [9].
The integration of AI into PDD workflows has created powerful new capabilities for analyzing complex biological systems and predicting compound activity, significantly accelerating the early stages of the drug discovery process [13].
Phenotypic Drug Discovery has firmly re-established itself as an essential approach for identifying first-in-class medicines with novel mechanisms of action. By focusing on therapeutic outcomes in physiologically relevant systems rather than predefined molecular targets, PDD has expanded the druggable target space to include previously inaccessible biological processes [2]. The continued evolution of PDD will be driven by advances in human-based phenotypic platforms, improved disease models, and sophisticated computational methods including AI and machine learning [8].
As these technologies mature and integrate, PDD is poised to address some of the most challenging limitations in drug discovery, particularly for complex diseases with polygenic origins or poorly understood biology. The future will likely see increased convergence of phenotypic and target-based approaches, creating hybrid workflows that leverage the strengths of both strategies [12]. This integration, powered by AI and multimodal data analysis, represents the next frontier in therapeutic discovery - enabling researchers to systematically navigate biological complexity while delivering innovative medicines for patients with unmet medical needs.
The history of modern drug discovery has been characterized by a pendulum swing between two fundamental strategies: Phenotypic Drug Discovery (PDD) and Target-Based Drug Discovery (TDD). PDD, the older of the two approaches, can be defined as a "compound-first" strategy that uses target-agnostic, system-based assays to identify pharmacologically active molecules based on their effects on disease phenotypes or translational biomarkers [16]. In contrast, TDD represents a "mechanism-first" approach focused on a specific molecular target—a gene product that provides a starting point for inventing a therapeutic which modulates its expression, function, or activity [16]. The evolution between these strategies represents more than a simple methodological preference; it reflects deeper philosophical differences in how researchers bridge the gap between understanding disease mechanisms and inventing effective medicines.
This analysis traces the historical trajectory of drug discovery from its phenotypic origins through the dominance of reductionist target-based approaches and the contemporary resurgence of phenotypic strategies, examining the technological and scientific forces driving these transitions and their implications for future therapeutic development.
The Historical Dominance of Phenotypic Drug Discovery
The empirical principles of PDD formed the foundation of early pharmaceutical development, with pioneers like Paul Ehrlich, who invented the first "magic bullet" (salvarsan) for syphilis from chemical dyes, and Sir James Black and Dr. Paul Janssen, who emphasized starting with a "pharmacologically active compound" [16]. George H. Hitchings Jr. highlighted the power of empirical, phenotypic screens when he stated in his 1988 Nobel lecture that "those early, untargeted studies led to the development of useful drugs for a wide variety of diseases and has justified our belief that this approach to drug discovery is more fruitful than narrow targeting" [16].
Before the genetic revolution, most medicines were identified primarily through this compound-first approach, relying on observable therapeutic effects in disease models or even serendipitous clinical observations rather than predefined molecular mechanisms. This empirical tradition produced many foundational therapeutics, but as molecular biology advanced, the limitations of this approach—including lengthy development cycles and uncertain mechanisms of action—became increasingly apparent, setting the stage for a paradigm shift.
The Ascendancy of Target-Based Drug Discovery
The genetic revolution of the 1980s-1990s, culminating in the sequencing of the human genome in 2001, fundamentally reshaped drug discovery philosophy [2]. The powerful new understanding of genes and their protein products created the vision that new medicines could be discovered rationally based on this molecular understanding of disease. This "mechanism-first" strategy promised greater efficiency, specificity, and a more scientific foundation for therapeutic development.
TDD dominated pharmaceutical research from approximately 1990-2010, driven by several perceived advantages:
- Clearer Development Pathways: Known molecular targets enabled rational drug design and optimization
- Improved Specificity: Drugs could be designed to interact specifically with validated targets, potentially reducing off-target effects
- High-Throughput Screening: Target-based assays were often more amenable to automation and miniaturization
- Biomarker Development: Known targets facilitated companion diagnostic development
The reductionist appeal of TDD aligned with the scientific zeitgeist of the period, leading to notable successes such as vemurafenib, a BRAF inhibitor for melanoma [16]. However, despite these advances, the cost of producing new medicines far outpaced the industry's ability to discover them, revealing a troubling gap between understanding disease mechanisms and actually inventing effective new medicines [16].
The Contemporary Resurgence of Phenotypic Approaches
A pivotal 2011 analysis by Swinney and Anthony of discovery strategies for new molecular entities approved by the FDA between 1999 and 2008 revealed a surprising pattern: a majority of first-in-class small-molecule drugs were discovered empirically through PDD approaches, while the majority of follower drugs were discovered using TDD [2] [16]. This analysis demonstrated that the mechanistic knowledge available when a program is initiated is often insufficient to provide a blueprint for discovering first-in-class medicines, creating a knowledge gap that PDD addresses empirically.
This revelation, combined with stagnating productivity in the pharmaceutical industry despite massive investments in target-based approaches, sparked a major resurgence in PDD beginning around 2011 [2]. Modern PDD has evolved significantly from its historical predecessors, now combining the original concept with sophisticated tools and strategies to systematically pursue drug discovery based on therapeutic effects in realistic disease models [2].
Table 1: Notable Drug Discoveries from Modern Phenotypic Approaches
| Drug | Disease Area | Key Target/Mechanism Identified | Screen Type |
|---|---|---|---|
| Ivacaftor, Tezacaftor, Elexacaftor [2] | Cystic Fibrosis | CFTR correctors/potentiators (channel folding/gating) | Cell lines expressing CFTR variants |
| Risdiplam, Branaplam [2] | Spinal Muscular Atrophy | SMN2 pre-mRNA splicing modulators | SMN2 reporter gene assays |
| Daclatasvir [2] | Hepatitis C | NS5A protein inhibitor | HCV replicon phenotypic screen |
| Lenalidomide [2] | Multiple Myeloma | Cereblon E3 ligase modulator (protein degradation) | Clinical observation (thalidomide derivatives) |
| SEP-363856 [2] | Schizophrenia | Novel mechanism (trace amine-associated receptor) | Phenotypic screen |
The return to phenotypic strategies has been facilitated by several technological advances:
- Improved Disease Models: Development of induced pluripotent stem cells (iPSCs), organoids, and more physiologically relevant cellular systems [16]
- Advanced Biomarkers: Sophisticated functional readouts with improved clinical translatability
- Chemical Biology Tools: Libraries designed for phenotypic screening with balanced diversity and tractability [16]
- Analytical Technologies: -omics approaches and bioinformatics for mechanism deconvolution
Quantitative Comparison: PDD vs. TDD Performance Metrics
Analyzing the relative performance of PDD and TDD approaches reveals distinct strengths and limitations for each strategy. The following table synthesizes data from industry analyses and clinical outcomes:
Table 2: Comparative Analysis of PDD vs. TDD Output and Characteristics
| Parameter | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| First-in-class medicines (1999-2008) [16] | Majority (from empirical discovery) | Minority |
| Follower medicines (1999-2008) [16] | Minority | Majority |
| Target Space | Novel, unexpected targets and mechanisms | Established, validated target classes |
| Mechanism of Action | Often identified post-discovery | Defined before compound optimization |
| Disease Models | Complex, systems-based, disease-relevant | Reductionist, target-focused |
| Typical Development Timeline | Often longer due to mechanism deconvolution | Potentially shorter with validated targets |
| Probability of Phase 2 → 3 Transition [16] | 32.4-48.6% (across strategies) | 32.4-48.6% (across strategies) |
| Probability of Phase 3 → Approval [16] | 50-59% (across strategies) | 50-59% (across strategies) |
| Major Challenge | Target identification, clinical translation | Target validation, clinical efficacy |
The data demonstrates that while TDD has proven effective for developing follower drugs that improve upon existing mechanisms, PDD has disproportionately contributed breakthrough first-in-class medicines with novel mechanisms of action. However, both approaches face significant challenges in late-stage development, with lack of therapeutic efficacy accounting for >50% of Phase 3 failures across strategies [16].
Experimental Framework: Methodologies for Modern PDD
Implementing a successful phenotypic drug discovery program requires carefully designed experimental workflows that balance physiological relevance with practical screening considerations. The following diagram illustrates a generalized PDD workflow:
Detailed Experimental Protocols
Phenotypic Screening Protocol: Cystic Fibrosis CFTR Correctors
The discovery of CFTR correctors (elexacaftor, tezacaftor) and potentiators (ivacaftor) for cystic fibrosis exemplifies modern PDD success [2].
Primary Screening Protocol:
- Cell Model: Utilize Fischer Rat Thyroid (FRT) cells co-expressing human CFTR mutants (e.g., ΔF508-CFTR) and a halide-sensitive yellow fluorescent protein (YFP)
- Assay Principle: Functional CFTR at membrane enables iodide influx, quenching YFP fluorescence
- Screening Format: 384-well plates, 10,000-100,000 compound libraries
- Compound Incubation: 24 hours to allow CFTR maturation and trafficking
- Assay Execution:
- Aspirate compound media and add iodide-free PBS
- Add iodide solution and measure YFP fluorescence (500/525 nm) every second for 20 seconds
- Calculate initial fluorescence slope as indicator of CFTR function
- Hit Criteria: Compounds showing >3 standard deviations above DMSO control mean
- Counterscreens: Toxicity assays, verification in primary human bronchial epithelial cells
Target Deconvolution Protocol: Mechanism of Action Studies
Following primary phenotypic screening, identifying molecular targets represents a critical PDD challenge.
Integrated Target Identification Workflow:
- Chemical Genetics:
- CRISPR/Cas9 knockout or RNAi screening with hit compounds
- Resistance generation and whole-exome sequencing of resistant clones
- Chemical Proteomics:
- Immobilize compound on solid support for affinity purification
- Incubate with cell lysates, wash, and elute bound proteins
- Identify proteins by mass spectrometry
- Transcriptomics/Proteomics:
- RNA sequencing or proteomic profiling of compound-treated vs. untreated cells
- Pattern matching to compounds with known mechanisms
- Bioinformatics Integration:
- Cross-reference multiple approaches to identify consensus targets
- Validate through genetic manipulation (overexpression/knockdown)
The Scientist's Toolkit: Essential Research Reagents for PDD
Implementing effective phenotypic screening requires carefully selected reagents and tools designed to maximize physiological relevance while maintaining screening feasibility.
Table 3: Essential Research Reagents for Modern Phenotypic Drug Discovery
| Reagent Category | Specific Examples | Function in PDD |
|---|---|---|
| Cell Models | iPSC-derived cells, primary human cells, organoids, 3D culture systems | Provide physiologically relevant environments for compound screening |
| Compound Libraries | Diversity-oriented synthesis libraries, known bioactives, natural product extracts | Source of chemical matter with balanced diversity and tractability [16] |
| Functional Reporters | Fluorescent calcium indicators, membrane potential dyes, YFP halide sensors | Enable measurement of functional phenotypes beyond simple viability |
| Genetic Tools | CRISPR/Cas9 libraries, siRNA collections, cDNA overexpression libraries | Facilitate target identification and validation |
| Analytical Technologies | High-content imagers, automated patch clamp, mass cytometers | Multiparametric readout capabilities for complex phenotypes |
| Biomarker Assays | Phospho-specific antibodies, metabolic flux assays, secreted protein markers | Bridge phenotypic observations to molecular mechanisms |
Integrated Approaches: The Future of Drug Discovery
The historical oscillation between PDD and TDD is evolving toward a more integrated approach that leverages the strengths of both strategies. The concept of "Mechanism-Informed PDD" (MIPDD) has emerged, which uses empirical assays to identify molecular mechanisms of action within target-based strategies [16]. This hybrid approach acknowledges that knowledge of a target alone does not always provide the molecular details required to predict a specific therapeutic response.
The future of drug discovery lies in recognizing that PDD and TDD represent complementary rather than competing approaches. PDD excels at identifying first-in-class medicines with novel mechanisms when knowledge gaps exist between targets and disease phenotypes, while TDD provides efficient optimization paths for validated targets and follower drugs. Successful organizations will maintain flexibility in selecting the optimal strategy based on the specific biological context, available tools, and project goals rather than adhering to methodological dogma.
The continued evolution of both approaches will be shaped by emerging technologies including artificial intelligence, functional genomics, and increasingly sophisticated disease models that further blur the traditional boundaries between phenotypic and target-based discovery, ultimately creating more opportunities to address unmet medical needs through innovative therapeutic mechanisms.
In the pharmaceutical landscape, two principal paradigms guide the discovery of new therapeutics: Target-Based Drug Discovery (TDD) and Phenotypic Drug Discovery (PDD). Historically, PDD was the primary method for discovering new medicines through observation of their effects on disease physiology in whole organisms or cellular models. The molecular biology revolution of the 1980s shifted focus toward TDD, a reductionist approach that modulates specific molecular targets with known roles in disease. Since approximately 2011, PDD has experienced a major resurgence following the observation that a majority of first-in-class drugs approved between 1999 and 2008 were discovered through phenotypic approaches without a predefined target hypothesis [2].
The modern iteration of PDD is defined by its focus on modulating a disease phenotype or biomarker in a realistic disease model, rather than a pre-specified target, to provide therapeutic benefit [2]. Conversely, TDD relies on an established causal relationship between a molecular target and a disease state. This technical guide examines the key rationales for choosing between these strategies, providing a structured decision-making framework for researchers and drug development professionals, supported by comparative data, experimental protocols, and practical toolkits.
Strategic Decision Framework: TDD vs. PDD
The choice between phenotypic and target-centric strategies depends on multiple project-specific variables, including the understanding of disease biology, desired innovation level, and available tools. The following table summarizes the key strategic considerations for selecting each approach.
Table 1: Strategic Decision Framework for TDD vs. PDD
| Decision Factor | Favor Phenotypic Screening (PDD) | Favor Target-Based Screening (TDD) |
|---|---|---|
| Target/Mechanism Understanding | No attractive or known target; complex, polygenic diseases with poorly understood pathophysiology [2] [17] | Well-validated target with established causal link to disease; understood mechanism of action [2] |
| Innovation Goals | First-in-class medicine; novel mechanism of action (MoA); expansion of druggable target space [2] | Best-in-class agent; improvement over existing therapies; optimization of known MoA [2] |
| Biological Complexity | Diseases requiring multi-target modulation (polypharmacology); unexpected biological connections [2] | Diseases with linear, well-defined pathways; single target modulation is sufficient for efficacy [2] |
| Technical Capabilities | Physiologically relevant disease models (e.g., human cell-based, microphysiological systems) [8] [18] | Target-based assay systems (e.g., enzymatic, binding, simple cellular assays) [17] |
| Risk Tolerance | Higher tolerance for uncertain target identity; investment in target deconvolution [17] [18] | Lower tolerance for target uncertainty; need for clear regulatory path based on target validation [17] |
Quantitative Performance Comparison
Empirical data reveals distinct performance patterns for TDD and PDD approaches in delivering new therapeutic agents. The following table summarizes key quantitative comparisons based on industry analyses.
Table 2: Performance Comparison of PDD and TDD Approaches
| Metric | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| First-in-Class Drugs | Disproportionate source of first-in-class medicines [2] | Less common origin for first-in-class drugs [2] |
| Approved Small-Molecule Drugs | Majority of approved small-molecule drugs originated from PDD approaches [19] | Only 123 of 1144 approved small-molecule drugs discovered by purely TDD methods [19] |
| Novel Mechanisms/Targets | Identifies unexpected cellular processes and novel mechanisms [2] | Primarily addresses known targets and established mechanisms |
| Target Identification | Requires subsequent target deconvolution - a key challenge [17] [18] | Target known from outset - no deconvolution needed |
| Integration Potential | Benefits from integration with TDD for mechanism elucidation [19] | Benefits from PDD data for understanding complex biology [19] |
Recent computational approaches demonstrate how integrating both strategies can enhance outcomes. The Knowledge-Guided Drug Relational Predictor (KGDRP) framework, which integrates multimodal biomedical data including biological networks, gene expression, and chemical structures within a heterogeneous graph structure, shows a 12% improvement in predictive performance in real-world screening scenarios and a 26% enhancement in target prioritization for drug target discovery [19].
Experimental Design and Methodologies
Phenotypic Screening Protocol Workflow
A robust phenotypic screening protocol requires careful model selection, assay development, and hit validation. The following diagram illustrates a generalized workflow for phenotypic screening campaigns:
The phenotypic screening workflow begins with careful definition of a disease-relevant phenotype, followed by selection of a physiological disease model that accurately recapitulates key aspects of human disease pathophysiology. Modern PDD increasingly utilizes human-based systems, including primary cells, induced pluripotent stem cells (iPSCs), and microphysiological systems (organ-on-a-chip) to enhance clinical translatability [8] [18]. After implementing a robust screening campaign, significant effort is dedicated to hit triage and prioritization, employing secondary assays and counter-screens to eliminate compounds with undesirable mechanisms [18]. A critical phase follows with target deconvolution to identify the molecular mechanism of action, employing methods such as affinity chromatography, expression cloning, protein microarrays, and biochemical suppression [20].
Integrated Graph-Based Learning Methodology
The KGDRP framework represents an advanced approach that integrates PDD and TDD data through biological heterogeneous graphs (BioHG). The following diagram illustrates this methodology:
The BioHG construction specifically incorporates several critical data types: drug response data (capturing drug-cell relationships), drug-target interaction data (describing drug-protein interactions), RNA expression profiles of cell lines (representing protein-cell line relationships), protein-protein interactions (from UniProt database), Gene Ontology data, and pathway data from Reactome [19]. Notably, drugs and cell lines are not directly connected in this graph structure, forcing the model to learn drug response through proteins, thereby enabling more comprehensive use of network information to enrich representations [19]. For the relationship between proteins and cell lines, proteins exhibiting expression values higher than the mean value of each cell line establish edges, with transcriptional expression values assigned as edge weights [19].
The framework incorporates several predictors: the RNA expression predictor, the drug-target interaction predictor, and the biological process predictor, which enable KGDRP to capture inherent correlations and dependencies across diverse biological networks through multi-task learning [19]. To address the drug cold-start problem (where drugs in PDD and TDD may not overlap), KGDRP introduces a transformation function that learns a mapping from chemical structure to knowledge-informed drug representations [19].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of either screening strategy requires specific research tools and reagents. The following table details essential components for establishing robust screening platforms.
Table 3: Essential Research Reagent Solutions for Drug Discovery Screening
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Cell Models | Primary human cells, iPSCs, engineered cell lines, co-culture systems | Provide physiologically relevant systems for phenotypic screening; engineered lines for target-based assays [8] [18] |
| Compound Libraries | Diverse small-molecule collections, targeted libraries, FDA-approved drug collections | Source of chemical matter for screening; diversity critical for PDD; focused libraries for TDD [2] |
| Detection Reagents | Fluorescent dyes, antibodies, luminescent probes, biosensors | Enable quantification of phenotypic changes or target engagement in screening assays [17] |
| Omics Tools | CRISPR libraries, transcriptomic profiling, proteomic arrays | Functional genomics for target identification/validation; mechanism of action studies [2] [17] |
| Specialized Assays | High-content imaging, reporter gene assays, pathway-specific assays | Phenotypic profiling and characterization; pathway modulation assessment in TDD [8] [17] |
Signaling Pathways and Biological Mechanisms
Phenotypic screening has uniquely expanded the "druggable target space" by identifying compounds that modulate previously unexplored biological pathways and mechanisms. The following diagram illustrates key pathways successfully targeted by PDD-derived therapeutics:
These successfully targeted pathways share a common characteristic: they would have been difficult to identify through purely target-based approaches. For example, the CFTR correctors that enhance protein folding and membrane insertion were discovered through phenotypic screens of cells expressing disease-associated CFTR variants, identifying compounds with an unexpected mechanism of action [2]. Similarly, the target and mechanism of lenalidomide were only elucidated several years post-approval, when it was found to bind the E3 ubiquitin ligase Cereblon and redirect its substrate selectivity [2].
The choice between target-centric and phenotype-centric strategies remains a fundamental consideration in therapeutic development. PDD offers distinct advantages for discovering first-in-class medicines with novel mechanisms, particularly for complex diseases with poorly understood pathophysiology. TDD provides a more direct path for optimizing known mechanisms and developing best-in-class agents against validated targets.
Future directions in the field point toward increased integration of both approaches through computational frameworks like KGDRP that leverage multimodal data [19], greater use of human-based physiological systems including microphysiological systems and organ-on-chip technologies [8] [18], and application of advanced artificial intelligence for target prediction and mechanism elucidation. The combination of robust phenotypic screening with modern target deconvolution technologies represents a powerful strategy for expanding the druggable genome and addressing unmet medical needs across diverse disease areas.
Ultimately, the decision between phenotypic and target-based approaches should be guided by specific project goals, biological understanding of the disease, and available technical resources rather than doctrinal adherence to either paradigm. Strategic integration of both approaches throughout the drug discovery pipeline offers the most promising path for delivering innovative therapeutics to patients.
Modern Workflows and Technologies: Implementing PDD and TDD in the AI Era
Target-based drug discovery (TDD) represents a cornerstone strategy in modern pharmaceutical research, operating in parallel and contrast to phenotypic drug discovery (PDD). While PDD identifies compounds based on their effects in complex biological systems without requiring prior knowledge of a specific molecular target, TDD follows a reductionist approach that begins with the selection and validation of a single molecular target believed to play a critical role in disease pathogenesis [21] [2]. This methodological dichotomy creates distinct advantages and challenges for each approach. TDD offers the significant benefit of a clear mechanism of action from the project's inception, facilitating rational drug design and optimization [22]. The TDD paradigm has been empowered by advances in genomics, structural biology, and screening technologies, enabling researchers to systematically pursue therapeutic interventions against a expanding array of biological targets [2] [22].
The disproportionate number of first-in-class medicines originating from PDD between 1999-2008 sparked renewed interest in phenotypic approaches [2]. However, TDD remains a dominant force in drug discovery, particularly for programs where the target biology is well-understood and the primary goal is to create best-in-class therapeutics against validated mechanisms rather than discover novel biology [2]. The TDD pipeline comprises a series of methodical stages, each with defined objectives and decision gates, designed to maximize the probability of clinical success while efficiently allocating resources. This technical guide provides an in-depth examination of the core TDD pipeline, from initial target identification through high-throughput screening, while contextualizing its strategic position within the broader drug discovery landscape that includes PDD approaches.
Phase 1: Target Identification and Validation
Principles of Target Identification
Target identification represents the foundational stage of the TDD pipeline, focusing on the selection of a biological entity whose modulation is expected to provide therapeutic benefit. A promising drug target typically exhibits several key properties [22]:
- A confirmed role in the pathophysiology of a disease and/or is disease-modifying
- Uneven expression distribution throughout the body to enable therapeutic targeting without widespread systemic effects
- An available 3D-structure to assess druggability through computational and experimental methods
- Being easily 'assayable' to enable high-throughput screening campaigns
- A promising toxicity profile where potential adverse effects can be predicted using phenotypic and bioinformatic data
The process of identifying a novel drug target can follow one of two principal strategic pathways (Figure 1) [22]. Target discovery operates on the paradigm that discovering a new drug requires first finding a new target, after which compound libraries are screened to identify molecules that interact with this target. In contrast, target deconvolution begins with a drug or compound that demonstrates efficacy, with the molecular target being identified retrospectively.
Figure 1: Strategic Pathways for Target Identification in TDD
Target Validation Techniques
Once a potential target is identified, rigorous validation is essential to demonstrate that modulating its activity will produce a therapeutic effect with an acceptable safety profile. Comprehensive target validation typically requires 2-6 months to complete and applies multiple complementary techniques to build compelling evidence for the biological target [23]. The three major components of target validation using human data include tissue expression profiling, genetic evidence, and clinical experience [24]. For each component, specific metrics can guide investment decisions and confidence levels (Table 1).
Table 1: Key Techniques for Target Validation
| Validation Category | Specific Techniques | Key Outputs/Metrics |
|---|---|---|
| Functional Analysis | In vitro assays using 'tool' compounds; Pharmacological modulation | Demonstration of desired biological effect; Dose-response relationships; Potency measurements (IC50, EC50) |
| Expression Profiling | mRNA and protein distribution analysis in healthy vs. disease states; qPCR; Immunohistochemistry | Correlation of target expression with disease progression; Tissue-specific expression patterns |
| Genetic Validation | Genome-wide association studies (GWAS); Genetic linkage analysis; siRNA/shRNA screening | Evidence of target-disease association from human genetics; Phenotypic effects of gene suppression |
| Biomarker Identification | Transcriptomics (qPCR); Protein analyte detection (Luminex); Flow cytometry | Quantifiable biomarkers for monitoring target engagement and therapeutic efficacy |
| Cell-Based Models | 3D cultures; Co-culture systems; Human induced pluripotent stem cells (iPSC) | Disease-relevant cellular models for evaluating target modulation in physiological context |
According to the National Academies, establishing pharmacologically relevant exposure levels and target engagement are two key steps in target validation [24]. Additionally, there is growing recognition of the importance of rapid target invalidation to avoid costly investment in targets that ultimately lack therapeutic potential. The ultimate validation occurs when a drug engaging the target demonstrates safety and efficacy in patients, but the goal of pre-clinical validation is to build sufficient confidence to justify proceeding to clinical development [24].
Phase 2: Assay Development and High-Throughput Screening
Fundamentals of High-Throughput Screening
High-throughput screening (HTS) serves as the primary engine for lead discovery in the TDD pipeline, enabling the rapid testing of hundreds of thousands to millions of compounds against a validated biological target [21] [25]. HTS leverages automation, miniaturization, and parallel processing to conduct biological or chemical tests on an unprecedented scale, dramatically accelerating the early drug discovery process [25] [26]. A typical HTS system consists of several integrated components: robotics for plate handling, liquid dispensing devices for reagent and compound transfer, environmental controllers for incubation, and sensitive detectors for signal readout [25].
The core labware for HTS is the microtiter plate, which features a grid of small wells arranged in standardized formats. Modern HTS primarily utilizes 384-well or 1536-well plates, with ongoing trends toward further miniaturization to 3456-well formats to reduce reagent costs and increase throughput [25] [26]. The working volumes in these systems have decreased substantially, with typical assays now running in 2.5-10 μL total volume, and ultra-high density systems operating with volumes as low as 1-2 μL per well [26]. This miniaturization enables the screening of vast compound libraries while conserving precious biological reagents and chemical compounds.
HTS assays are predominantly classified as either biochemical (cell-free) or cell-based formats. Biochemical assays measure direct interactions between compounds and purified targets (enzymes, receptors), while cell-based assays examine compound effects in a more physiological context, including pathway activation and phenotypic changes [21]. The choice between these formats depends on the target biology, assay feasibility, and the desired information about compound activity.
HTS Assay Development and Quality Control
Robust assay development is critical for successful HTS campaigns. A well-developed HTS assay must balance sensitivity, reproducibility, and scalability while maintaining biological relevance [21]. The development process involves optimizing reagent concentrations, incubation times, detection methods, and tolerance to dimethyl sulfoxide (DMSO)—the common solvent for compound libraries.
Several key performance metrics are employed to ensure assay quality and reliability (Table 2). The Z'-factor is particularly important, providing a normalized measure of assay robustness that accounts for both the signal dynamic range and data variation. A Z'-factor between 0.5 and 1.0 indicates an excellent assay suitable for HTS [21]. Other critical parameters include the signal-to-noise ratio, signal window, and coefficient of variation across wells and plates.
Table 2: Key Performance Metrics for HTS Assay Validation
| Performance Metric | Calculation/Definition | Acceptance Criteria | Application in HTS |
|---|---|---|---|
| Z'-factor | 1 - (3σpositive + 3σnegative)/|μpositive - μnegative| | 0.5-1.0: Excellent assay0-0.5: Marginal assay<0: Poor assay | Overall assay quality assessment; Day-to-day robustness |
| Signal-to-Background Ratio | Meansignal / Meanbackground | >3: Typically acceptableHigher values preferred | Measures assay window magnitude |
| Signal-to-Noise Ratio | (Meansignal - Meanbackground) / σ_background | >10: Typically acceptableDependent on assay type | Assesses detection sensitivity |
| Coefficient of Variation (CV) | (σ / μ) × 100% | <10-20% depending on assay type | Measures well-to-well reproducibility |
| Strictly Standardized Mean Difference (SSMD) | (μpositive - μnegative) / √(σ²positive + σ²negative) | >3: Strong hit selection | Hit selection in screens with replicates |
Recent advances in HTS include the development of quantitative HTS (qHTS) paradigms, where compounds are tested at multiple concentrations to generate concentration-response curves directly from the primary screen [25] [27]. This approach provides richer pharmacological data early in the discovery process, enables better assessment of structure-activity relationships, and reduces false positive and negative rates by more fully characterizing compound effects.
The HTS workflow follows a staged approach to efficiently identify high-quality hits (Figure 2). The process begins with primary screening of entire compound libraries, typically in single-point format, to identify initial "hits." These hits progress to confirmation screening, often with replicates and counter-screens to eliminate false positives, followed by concentration-response experiments to determine compound potency (IC50/EC50 values).
Figure 2: HTS Workflow from Assay Development to Hit Identification
The Scientist's Toolkit: Essential Research Reagents and Technologies
Successful implementation of the TDD pipeline requires a comprehensive suite of research tools and technologies. The following table details essential reagents and platforms used throughout target validation and HTS phases.
Table 3: Essential Research Reagents and Technologies for TDD
| Tool Category | Specific Examples | Key Applications in TDD |
|---|---|---|
| Gene Modulation Tools | siRNA, shRNA, CRISPR-Cas9 | Target validation through gene knockdown/knockout; Functional genomics |
| Detection Technologies | Fluorescence Polarization (FP), TR-FRET, Fluorescence Intensity, Luminescence | HTS assay detection; Quantifying biochemical interactions and cellular responses |
| Cell-Based Model Systems | Immortalized cell lines, Primary cells, iPSC-derived cells, 3D cultures, Co-culture systems | Disease-relevant models for target validation and phenotypic screening |
| Compound Libraries | Diverse small molecule collections, Focused libraries, Natural product extracts | Source of chemical starting points for HTS campaigns |
| Automation & Robotics | Liquid handlers, Plate readers, Automated incubators, Central robotics systems | Enabling HTS throughput and reproducibility; Reducing manual labor |
| Labeling & Detection Reagents | Fluorescent probes, Antibodies, Aptamers, Luminescent substrates | Signal generation in HTS assays; Target detection and quantification |
| Bioinformatic Tools | Chemical databases, Structural modeling software, Data analysis pipelines | Target assessment; Compound library design; HTS data analysis and hit selection |
Strategic Considerations: TDD vs. PDD in Modern Drug Discovery
The choice between TDD and PDD approaches represents a fundamental strategic decision in drug discovery programming. Each paradigm offers distinct advantages and faces particular challenges (Table 4). TDD provides a clear mechanism of action from the outset, enables rational drug design based on target structure, typically offers higher throughput in screening, and facilitates the development of pharmacodynamic biomarkers for clinical development [22]. Conversely, PDD has demonstrated a superior track record in producing first-in-class medicines with novel mechanisms, expands the "druggable target space" to include previously unexplored biological processes, and identifies compounds that act through polypharmacology (simultaneous modulation of multiple targets) [2].
Table 4: Comparative Analysis of TDD and PDD Approaches
| Parameter | Target-Based Drug Discovery (TDD) | Phenotypic Drug Discovery (PDD) |
|---|---|---|
| Starting Point | Known molecular target with hypothesized role in disease | Disease-relevant phenotype or biomarker without pre-specified target |
| Mechanism of Action | Known from project inception | Often unknown initially; requires deconvolution |
| Throughput Potential | Typically higher; streamlined assay systems | Often lower due to complex assay systems |
| Druggable Space | Limited to targets with established assay feasibility | Expands to novel targets and mechanisms |
| Historical Success | Majority of best-in-class drugs | Disproportionate number of first-in-class drugs |
| Target Identification | Required before screening | Required after hit identification |
| Chemical Optimization | Facilitated by structural knowledge of target | Often empirical without structural guidance |
| Clinical Translation | Biomarker strategies can be developed early | Physiological relevance may improve translation |
Rather than viewing TDD and PDD as competing strategies, modern drug discovery increasingly recognizes their complementary nature [22]. Many successful drug discovery programs employ elements of both approaches—using phenotypic assays to validate target biology and assess compound efficacy in physiologically relevant systems, while employing target-based assays for mechanistic studies and structure-based optimization. The strategic integration of both paradigms represents a powerful approach to addressing the ongoing challenges of drug discovery productivity.
Recent trends include the use of human-based phenotypic platforms throughout the discovery process for hit triage and prioritization, elimination of hits with unsuitable mechanisms, and supporting clinical strategies through pathway-based decision frameworks [8]. As these approaches mature, they offer the potential to generate better leads faster by leveraging the strengths of both TDD and PDD within integrated discovery workflows.
Phenotypic Drug Discovery (PDD) has re-emerged as a critical strategy in modern therapeutic development, driven by the observation that it disproportionately yields first-in-class medicines [2]. Unlike Target-Based Drug Discovery (TDD), which begins with a predefined molecular hypothesis, PDD identifies compounds based on their ability to modify disease-relevant phenotypes in biologically complex systems without prior knowledge of the specific drug target [2] [17]. This approach has successfully addressed complex diseases where the underlying pathophysiology is incompletely understood or where multi-target modulation provides therapeutic benefits [2]. Modern PDD combines this foundational concept with advanced tools and strategies, systematically pursuing drug discovery based on therapeutic effects in realistic disease models [2]. This technical guide details the core components of the PDD pipeline, from assay design principles to hit identification and validation, providing researchers with a framework for implementing this powerful approach.
The distinction between PDD and TDD represents more than a technical difference; it fundamentally shapes discovery strategy and outcomes. Between 1999 and 2008, an analysis revealed that a majority of first-in-class drugs were discovered through phenotypic approaches rather than target-based methods [2] [19]. PDD expands the "druggable target space" to include unexpected cellular processes and novel mechanisms of action (MoA), as demonstrated by breakthroughs in cystic fibrosis, spinal muscular atrophy, and hepatitis C treatment [2]. Furthermore, PDD naturally accommodates and even exploits polypharmacology – where a compound engages multiple targets – which can be advantageous for treating complex, polygenic diseases [2]. For broader adoption, key challenges need resolution, including the progression of poorly qualified leads and the advancement of compounds with undesirable mechanisms that fail at later stages [8].
Phenotypic Assay Design Fundamentals
Core Principles and System Selection
Effective phenotypic assays balance biological relevance with technical feasibility, requiring careful consideration of multiple factors:
- Disease Relevance: The assay system must capture key aspects of human disease pathophysiology. This includes relevant cell types, disease-associated stimuli, and endpoints that reflect clinical manifestations [17].
- Translational Bridge: Establish a "chain of translatability" connecting the assay phenotype to human disease biology through 'omics signatures and pathway engagement [17].
- Technical Robustness: Ensure the assay meets standard performance criteria (Z' factor >0.5, coefficient of variation <20%) to reliably detect compound effects amid system complexity [17].
- Scalability: Design with screening feasibility in mind, considering timeline, resource requirements, and compatibility with automation when moving to higher-throughput formats [8].
Modern phenotypic screening uses biological systems directly for new drug screening, ranging from cell-based setups to higher-order screening using small animal models [28]. The choice of experimental model represents a critical decision point that balances physiological relevance with practical screening constraints.
Table 1: Comparison of Phenotypic Screening Models
| Model System | Physiological Relevance | Throughput Capacity | Key Applications | Major Limitations |
|---|---|---|---|---|
| 2D Cell Cultures | Moderate | High | Initial hit identification, mechanism studies | Limited tissue context, simplified microenvironment |
| 3D Organoids/Spheroids | High | Medium | Complex cell-cell interactions, tissue morphogenesis | Higher variability, more complex image analysis |
| Microphysiological Systems (Organs-on-Chips) | High | Low-medium | Human pathophysiology, complex tissue interfaces | Specialized equipment, limited throughput |
| Small Animal Models | Highest | Low | Whole-organism physiology, integrated systems | Low throughput, high cost, translatability questions |
Key Technological Components
Advanced technologies enable the detailed interrogation of complex phenotypes in modern PDD:
- High-Content Imaging and Analysis: Multiparametric imaging captures morphological and spatial information at single-cell resolution, providing rich datasets on compound effects [28]. Automated image analysis pipelines extract quantitative features that define phenotypic states.
- Functional Genomic Integration: CRISPR-based screening identifies genes and pathways that modulate disease phenotypes, validating targets and generating mechanistic insights [17].
- Transcriptomic Profiling: Gene expression signatures contextualize compound effects within known disease pathways and enable comparison to reference compounds [17] [19].
- Microphysiological Systems: These human cell-based platforms recapitulate tissue-level and organ-level functions, providing more physiologically relevant contexts for compound testing [8].
Quantitative Phenotypic Endpoints and Assay Validation
The selection and validation of quantitative endpoints is fundamental to successful phenotypic screening. Modern approaches move beyond single-parameter measurements to capture multidimensional phenotypes that better reflect disease biology.
Table 2: Categories of Phenotypic Endpoints and Their Applications
| Endpoint Category | Measured Parameters | Detection Methods | Therapeutic Area Examples |
|---|---|---|---|
| Morphological | Cell size, shape, organelle distribution, spatial relationships | High-content imaging, automated microscopy | Oncology, neurodegenerative diseases |
| Proteomic | Protein expression, localization, post-translational modifications | Immunofluorescence, FRET, flow cytometry | Immunology, inflammation |
| Functional | Calcium flux, membrane potential, metabolic activity | FLIPR, electrophysiology, Seahorse analyzer | Cardiology, metabolic diseases |
| Secretory | Cytokine release, hormone secretion, extracellular matrix deposition | ELISA, luminescence, mass spectrometry | Immunology, fibrosis |
| Transcriptional | Gene expression changes, pathway activation | Reporter gene assays, RT-qPCR | Oncology, virology |
Assay validation establishes the reliability and predictive value of the phenotypic system. The "Phenotypic Screening Rule of 3" provides a framework for this process, emphasizing three critical elements: (1) clinical relevance of the assay system, (2) pharmacological credibility of known reference compounds, and (3) statistical robustness of the assay performance [17]. Technical validation should establish a Z' factor >0.5, signal-to-noise ratio >3, and coefficient of variation <20% for key parameters. Biological validation should demonstrate that the assay detects efficacy of known therapeutic agents with appropriate potencies and generates disease-relevant phenotypes that align with clinical manifestations.
Experimental Workflow for Phenotypic Screening
The following diagram illustrates the core workflow for implementing a phenotypic screening campaign, from model establishment through hit identification:
Model System Establishment
Initiate the PDD pipeline by selecting and validating a disease-relevant biological system:
- Cell Line Selection: Choose disease-relevant cell types, preferably primary human cells or patient-derived induced pluripotent stem cells (iPSCs) that capture key disease pathophysiology [8]. Consider incorporating multiple cell types to model tissue-level interactions.
- Disease Modeling: Introduce disease-specific perturbations through genetic manipulation (e.g., CRISPR), disease-associated stimuli (e.g., inflammatory cytokines), or patient-derived materials. Ensure the model produces a measurable phenotype relevant to the clinical condition.
- System Characterization: Thoroughly profile the model using transcriptomic, proteomic, and functional analyses to establish its relationship to human disease biology [17].
Assay Implementation and Screening
Execute the phenotypic screen with appropriate controls and quality metrics:
- Library Design: Curate diverse compound collections that maximize chemical and target space exploration. Include known tool compounds as pharmacological anchors for mechanism interpretation.
- Automation and Scaling: Implement robotic liquid handling and high-content imaging systems for consistent assay execution. Maintain physiological relevance while achieving necessary throughput.
- Quality Control: Include reference controls on every plate to monitor assay performance. Establish statistical thresholds for hit calling based on control compound performance and biological variability.
Hit Triage and Prioritization Strategies
The following workflow outlines the multi-parameter approach required for effective hit triage in phenotypic screening:
Following primary screening, hit triage eliminates artifacts and prioritizes compounds with desirable properties:
- Potency and Efficacy Confirmation: Retest hits in concentration-response format to confirm activity and determine half-maximal effective concentration (EC50) and maximal response (efficacy) values.
- Counter-Screening: Eliminate false positives and pan-assay interference compounds (PAINS) through orthogonal assays that assess general cytotoxicity, fluorescence interference, and assay-specific artifacts.
- Chemical Assessment: Evaluate compound properties, including structural integrity (LC-MS confirmation), chemical novelty, and developability based on physicochemical properties.
- Phenotypic Specificity: Assess compound effects in related but distinct phenotypic assays to establish selectivity for the disease-relevant phenotype.
- Early Mechanistic Exploration: Use functional genomics (CRISPR) or chemoproteomics to begin investigating potential mechanisms while maintaining target-agnostic positioning.
Computational approaches are increasingly valuable for hit prioritization. Methods like the Knowledge-Guided Drug Relational Predictor (KGDRP) integrate multimodal biomedical data, including biological network data, gene expression data, and chemical structures within a heterogeneous graph framework to enhance prediction accuracy and provide biological context for screening hits [19].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful implementation of phenotypic screening requires carefully selected reagents and platforms that maintain biological relevance while enabling robust detection.
Table 3: Essential Research Reagents and Platforms for Phenotypic Screening
| Reagent/Platform Category | Specific Examples | Key Function in PDD | Technical Considerations |
|---|---|---|---|
| Complex Cell Models | Patient-derived iPSCs, 3D organoids, co-culture systems | Provide disease-relevant cellular context with appropriate pathophysiology | Batch-to-batch variability, characterization depth, scalability limitations |
| Biosensors and Reporters | GFP-tagged proteins, calcium indicators, FRET-based biosensors | Enable dynamic monitoring of pathway activation and cellular responses | Potential perturbation of native biology, technical validation requirements |
| High-Content Imaging Reagents | Multiplexable fluorescent dyes, antibodies, viability indicators | Facilitate multiparametric readouts of complex phenotypes | Spectral overlap, photostability, compatibility with live-cell imaging |
| Functional Genomic Tools | CRISPR libraries, RNAi collections, cDNA overexpression sets | Enable systematic perturbation to identify targets and mechanisms | Delivery efficiency, off-target effects, interpretation complexity |
| Microphysiological Systems | Organ-on-chip platforms, 3D bioprinted tissues, perfusion systems | Model tissue-level structure and function with improved physiology | Throughput limitations, specialized expertise requirements, cost |
| Computational Integration Tools | KGDRP, network analysis, image analysis pipelines | Integrate multimodal data, prioritize hits, elucidate mechanisms | Data heterogeneity, computational resource requirements, interpretability |
Mechanism Elucidation and Target Deconvolution
While not always required for compound advancement, understanding mechanism of action (MoA) provides valuable insights for safety profiling and clinical development. Modern approaches to target deconvolution include:
- Chemical Biology Methods: Employ affinity purification using compound analogs, activity-based protein profiling, or photoaffinity labeling to identify cellular binding partners [2].
- Functional Genomics: Combine genome-wide CRISPR screening with compound treatment to identify genes that modulate sensitivity or resistance to phenotypic hits [17].
- Transcriptomic Profiling: Compare compound-induced gene expression signatures to reference databases (e.g., Connectivity Map) to infer mechanism and predict potential toxicity [17].
- Multiparametric Phenotypic Profiling: Collect high-dimensional phenotypic data and compare to compounds with known mechanisms to identify similar profiles [8].
Notably, the field is shifting perspective that comprehensive target identification may not be essential for all phenotypic-derived compounds, particularly if the phenotypic efficacy and selectivity are well-established and the compound demonstrates acceptable safety margins [2] [8].
Integration with Target-Based Approaches
The most productive drug discovery strategies leverage both phenotypic and target-based approaches. Computational frameworks like KGDRP demonstrate that integrating PDD and TDD data can enhance both drug response prediction and target identification [19]. This integration creates a virtuous cycle where phenotypic observations inform target validation, and target knowledge enhances phenotypic assay design. The emerging paradigm treats PDD and TDD as complementary rather than competing approaches, recognizing that each brings distinct strengths to addressing different aspects of the drug discovery process.
KGDRP exemplifies this integration by incorporating biological network data, gene expression data, and chemical structures within a heterogeneous graph, enabling simultaneous prediction of drug response and potential targets [19]. This approach addresses the "cold-start" problem where drugs in phenotypic screens may lack target annotations, thereby bridging the gap between phenotypic observations and mechanistic understanding.
PDD represents a powerful approach for identifying first-in-class therapies, particularly for complex diseases with poorly understood pathophysiology. By focusing on disease-relevant phenotypes in biologically complex systems, PDD expands the druggable genome and enables discovery of novel mechanisms of action. Successful implementation requires careful attention to assay design, model selection, hit triage, and mechanism elucidation. As human-based phenotypic platforms, computational integration methods, and mechanistic tools continue to advance, PDD is poised to deliver an increasing number of transformative medicines. The future of phenotypic discovery lies not in replacing target-based approaches, but in strategic integration that leverages the complementary strengths of both paradigms.
The integration of artificial intelligence (AI) and machine learning (ML) is fundamentally reshaping the landscape of drug discovery, bridging the historical divide between phenotypic drug discovery (PDD) and target-based drug discovery (TDD). Historically, PDD has contributed to a larger proportion of first-in-class medicines by observing compound effects in complex biological systems without requiring prior knowledge of a specific molecular target, while TDD has enabled rational drug design based on well-defined molecular mechanisms [29] [9]. This traditional dichotomy is now being transcended by hybrid approaches that leverage computational modeling, artificial intelligence, and multi-omics technologies to create integrated workflows that enhance therapeutic efficacy and overcome resistance [29]. AI technologies have evolved from experimental curiosities to clinically valuable tools, with AI-designed therapeutics now progressing through human trials across diverse therapeutic areas, representing nothing less than a paradigm shift that replaces labor-intensive, human-driven workflows with AI-powered discovery engines capable of dramatically compressing timelines and expanding chemical and biological search spaces [15].
The global machine learning in drug discovery market is experiencing significant expansion, with projections indicating substantial revenue growth through 2034, driven by emerging trends and strong demand across key sectors [30]. This growth is catalyzed by AI's ability to analyze massive datasets, identify complex patterns, and generate novel hypotheses at scales and speeds impossible for human researchers alone. By leveraging algorithmic models that learn from large datasets to identify patterns, predict outcomes, and make data-driven decisions across the drug discovery process, ML accelerates early-stage research, enables identification of novel drug candidates, enhances target validation, optimizes lead compounds, and predicts toxicity and pharmacokinetics, thereby significantly reducing both R&D time and cost [30]. This technical guide examines how these computational approaches are accelerating target prediction and data analysis within both PDD and TDD frameworks, providing researchers and drug development professionals with practical methodologies and insights for implementation.
AI-Driven Methodologies for Phenotypic and Target-Based Discovery
AI-Enhanced Phenotypic Drug Discovery
Phenotypic drug discovery has experienced a renaissance in recent years, growing from less than 10% to an estimated 25-40% of the project portfolios in major pharmaceutical companies like AstraZeneca and Novartis between 2012 and 2022 [9]. This resurgence is largely attributable to AI and ML technologies that have overcome traditional limitations of phenotypic approaches. Modern AI tools enable automated analysis of complex cell image data, extraction of diverse morphological features, and clustering of cellular phenotypes to identify potential drug candidates with unprecedented efficiency [9]. These computational methods leverage multimodal data integration, combining chemical structure features with extracted image features to elucidate Mode of Action (MoA) and bioactivity properties with significantly improved prediction power [9].
Advanced computational platforms like Sonrai Discovery exemplify this approach by integrating complex imaging, multi-omic, and clinical data into a single analytical framework. Their platform employs foundation models trained on thousands of histopathology and multiplex imaging slides to extract features and identify novel biomarkers that can be linked to clinical outcomes [31]. This AI-driven phenotypic profiling enables researchers to uncover links between molecular features and disease mechanisms more rapidly than traditional methods. The transparency of these workflows is crucial—using trusted and tested tools within trusted research environments allows clients to verify exactly what data goes in and what insights come out, building essential confidence in AI-generated results [31].
The success of AI-enhanced PDD is evidenced by recently approved treatments identified through these methods. Vamorolone for Duchenne muscular dystrophy and Risdiplam for spinal muscular atrophy both emerged from phenotypic approaches that would have been unlikely to succeed through traditional target-based methods [9]. These drugs target pathways and mechanisms that lacked well-characterized functional roles in disease, making them elusive targets for conventional approaches. Similarly, Daclatasvir (Daklinza) for hepatitis C was identified through phenotypic screening and later found to target NS5A, a non-structural protein with no enzymatic activity that had remained an elusive target for many years [9].
AI-Optimized Target-Based Drug Discovery
Target-based drug discovery has been equally transformed by AI and ML methodologies, particularly through the application of graph neural networks, generative chemistry, and physics-enabled design strategies. These approaches enable researchers to move beyond single-target paradigms to address complex disease networks and pathways. Tools like PDGrapher, developed by researchers at Harvard Medical School, represent this new generation of target discovery platforms [32]. This AI model focuses on identifying multiple drivers of disease in cells and predicts therapies that can restore cells to healthy function, moving away from traditional approaches that test one protein target or drug at a time [32].
PDGrapher operates as a graph neural network that doesn't just examine individual data points but maps the relationships between genes, proteins, and signaling pathways inside cells to predict optimal combination therapies that correct underlying cellular dysfunction [32]. The model is trained on datasets of diseased cells before and after treatment, learning which genes to target to shift cells from diseased to healthy states. In validation tests across 19 datasets spanning 11 cancer types, the tool accurately predicted known drug targets that had been deliberately excluded during training, while also identifying additional candidates supported by emerging evidence [32]. The system demonstrated superior accuracy and efficiency compared to similar tools, ranking correct therapeutic targets up to 35% higher than other models and delivering results up to 25 times faster [32].
Leading AI-driven drug discovery platforms exemplify the successful implementation of these approaches in TDD. Exscientia's end-to-end platform integrates AI at every stage from target selection to lead optimization, using deep learning models trained on vast chemical libraries and experimental data to propose novel molecular structures that satisfy precise target product profiles [15]. Similarly, Schrödinger's physics-enabled design strategy, exemplified by the advancement of the TYK2 inhibitor zasocitinib (TAK-279) into Phase III clinical trials, demonstrates how computational approaches can successfully advance compounds through late-stage clinical testing [15].
Hybrid PDD-TDD Integration Using AI
The most significant advancement in AI-driven drug discovery is the emergence of hybrid approaches that seamlessly integrate phenotypic and target-based strategies. The 2025 acquisition of Exscientia by Recursion Pharmaceuticals in a $688 million merger exemplifies this trend, combining Exscientia's strength in generative chemistry and design automation with Recursion's extensive phenomics and biological data resources to create an integrated "AI drug discovery superpower" [15]. Such integrated platforms leverage the target-agnostic advantage of PDD with the mechanistic precision of TDD, creating synergistic workflows that overcome the limitations of either approach alone.
These hybrid systems employ closed-loop design-make-test-learn cycles powered by cloud infrastructure and foundation models. For instance, Exscientia's integrated AI-powered platform built on Amazon Web Services links its generative-AI "DesignStudio" with a UK-based "AutomationStudio" that uses state-of-the-art robotics to synthesize and test candidate molecules [15]. This creates a continuous feedback loop where phenotypic data informs target identification, and target-based design generates compounds for phenotypic validation. The resulting workflows enable companies to rapidly iterate through compound design and testing phases, with Exscientia reporting in silico design cycles approximately 70% faster and requiring 10x fewer synthesized compounds than industry norms [15].
AI models like PDGrapher further facilitate this integration by identifying cause-effect biological drivers of disease, helping researchers understand why certain drug combinations work—offering new biological insights that propel biomedical discovery forward [32]. This approach is particularly valuable for complex diseases fueled by multiple pathways, such as cancer, where tumors can develop resistance to drugs targeting single mechanisms. By identifying multiple targets involved in disease progression, AI-enabled hybrid approaches help circumvent this persistent challenge in drug development [32].
Experimental Protocols and Methodologies
Protocol: High-Content Phenotypic Screening with AI Analysis
Purpose: To identify novel therapeutic compounds and their mechanisms of action through automated analysis of compound-induced phenotypic changes in disease-relevant cell models.
Materials and Reagents:
- 3D Cell Culture Systems: Primary cells or cell lines relevant to disease pathology (e.g., patient-derived organoids) [31]
- MO:BOT Platform or equivalent automation system: For standardized 3D cell culture handling, including automated seeding, media exchange, and quality control [31]
- Multiplex Assay Kits: For measuring multiple parameters simultaneously (cell viability, apoptosis, mitochondrial function, etc.)
- High-Content Imaging System: Confocal or fluorescent microscopy capable of automated multi-well plate scanning
- AI-Powered Image Analysis Software: Such as Ardigen's phenAID platform or equivalent for extraction of morphological features [9]
Procedure:
- Platform Setup: Prepare 3D cell cultures using automated systems like the MO:BOT platform, which standardizes organoid production and rejects sub-standard organoids before screening to ensure reproducibility [31].
- Compound Treatment: Treat cells with compound libraries across concentration gradients, including appropriate controls (vehicle, positive, and negative controls).
- Multiparametric Data Collection: Using high-content imaging systems, capture temporal and spatial data on multiple cellular features including morphology, protein localization, and organelle structure.
- Feature Extraction: Apply ML algorithms to extract quantitative morphological features from cell images, creating "phenotypic fingerprints" for each treatment condition [9].
- Pattern Recognition: Use unsupervised learning methods (clustering, dimensionality reduction) to group compounds with similar phenotypic effects and identify novel activity patterns.
- Mechanism Prediction: Integrate phenotypic profiles with chemical structure data using multimodal AI approaches to predict potential mechanisms of action [9].
- Validation: Confirm predictions through targeted genetic or pharmacological experiments in relevant disease models.
AI Integration Points:
- Implement foundation models pre-trained on thousands of histopathology and multiplex imaging slides to identify novel biomarkers [31]
- Apply transfer learning to adapt general models to specific disease contexts
- Use generative AI to suggest chemical modifications that enhance desired phenotypic effects
Protocol: Graph Neural Network for Multi-Target Identification
Purpose: To identify optimal single or combination drug targets that reverse disease states at the cellular level using causal inference modeling.
Materials and Reagents:
- Transcriptomic Datasets: Bulk or single-cell RNA sequencing data from diseased and healthy control tissues
- Public Knowledge Bases: Curated pathway databases (KEGG, Reactome), protein-protein interaction networks, and drug-target databases
- Computational Resources: High-performance computing cluster with GPU acceleration for graph neural network training
- PDGrapher or Equivalent Software: Graph neural network platform for causal target identification [32]
Procedure:
- Network Construction:
- Build a heterogeneous knowledge graph integrating:
- Gene regulatory networks from transcriptomic data
- Protein-protein interaction networks from public databases
- Drug-target interactions from pharmacological databases
- Signaling pathways from curated knowledge bases
- Build a heterogeneous knowledge graph integrating:
Node Representation:
- Apply graph embedding algorithms to create numerical representations for each node (genes, proteins, compounds) in the network
- Incorporate multiple data modalities including sequence, structure, and functional annotation
Causal Inference:
- Train the graph neural network on datasets of diseased cells before and after effective treatments to learn patterns associated with therapeutic reversal of disease states [32]
- Implement attention mechanisms to identify the most influential nodes in the network for reversing disease phenotypes
Target Prioritization:
- Use the trained model to simulate interventions on potential targets and rank them by their predicted ability to shift cells from diseased to healthy states [32]
- Identify synergistic target combinations that show enhanced efficacy compared to single targets
Experimental Validation:
- Test top-ranked targets using CRISPR-based gene editing or RNA interference in disease-relevant cellular models
- Validate predicted combination therapies in appropriate animal models of disease
Validation Metrics:
- Benchmark against known effective treatments excluded from training data [32]
- Compare prediction accuracy with alternative methods (PDGrapher demonstrated 35% higher accuracy and 25x faster performance than comparable approaches) [32]
Data Presentation and Analysis
Quantitative Analysis of AI in Drug Discovery
Table 1: Machine Learning in Drug Discovery Market Analysis (2024-2034 Projection)
| Category | 2024 Market Share | Projected Growth | Key Drivers |
|---|---|---|---|
| By Application Stage | |||
| Lead Optimization | ~30% | Stable dominance | AI-driven optimization of drug efficiency, safety, and development timelines [30] |
| Clinical Trial Design & Recruitment | Emerging segment | Fastest CAGR (2025-2034) | Personalized trial models and biomarker-based stratification from patient data [30] |
| By Algorithm Type | |||
| Supervised Learning | 40% | Mature segment | Ability to predict drug activity using labeled datasets [30] |
| Deep Learning | Growing segment | Fastest growth period | Structure-based predictions and AlphaFold use in protein modeling [30] |
| By Therapeutic Area | |||
| Oncology | 45% | Maintained dominance | Rising cancer prevalence demanding personalized therapies [30] |
| Neurological Disorders | Emerging segment | Fastest CAGR | Growing incidences of Alzheimer's and Parkinson's [30] |
| By Region | |||
| North America | 48% | Stable growth | Substantial funding and FDA regulatory support for AI applications [30] |
| Asia Pacific | Growing segment | Fastest growing region | Abundant biological data and robust IT infrastructure [30] |
Table 2: Leading AI-Driven Drug Discovery Platforms and Their Clinical Progress (2025)
| Company/Platform | Core AI Technology | Therapeutic Areas | Clinical Stage Progress |
|---|---|---|---|
| Exscientia | Generative chemistry, Centaur Chemist approach | Oncology, Immunology, Inflammation | Eight clinical compounds designed; CDK7 inhibitor (GTAEXS-617) in Phase I/II; LSD1 inhibitor (EXS-74539) Phase I [15] |
| Insilico Medicine | Generative adversarial networks (GANs) | Idiopathic pulmonary fibrosis, Oncology | ISM001-055 (TNK inhibitor) showed positive Phase IIa results in IPF; target-to-clinic timeline of 18 months [15] |
| Schrödinger | Physics-based computational platform | Autoimmune diseases, Oncology | TYK2 inhibitor zasocitinib (TAK-279) advanced to Phase III trials [15] |
| Recursion | Phenomics-first approach, high-content screening | Rare diseases, Oncology | Merger with Exscientia created integrated phenomics-generative chemistry platform [15] |
| BenevolentAI | Knowledge-graph driven target discovery | Immunology, Neurology | Multiple candidates in clinical stages from knowledge-graph approach [15] |
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents and Platforms for AI-Driven Drug Discovery
| Research Tool | Function | Application in AI Workflows |
|---|---|---|
| MO:BOT Platform | Automated 3D cell culture system for standardizing organoid production | Generates reproducible, human-relevant tissue models for phenotypic screening; rejects sub-standard organoids before screening [31] |
| Ardigen phenAID Platform | AI-powered analysis of high-content screening datasets | Reduces analysis time and enhances prediction quality for HCS datasets; enables phenotypic profiling [9] |
| Sonrai Discovery Platform | Integrated analysis of imaging, multi-omic and clinical data | Provides trusted research environment with transparent AI pipelines; applies foundation models to extract features from imaging data [31] |
| Cenevo/Labguru AI Assistant | Data management and AI integration platform | Embeds intelligent tools directly into scientific software; supports smarter search, experiment comparison, and workflow generation [31] |
| Nuclera eProtein Discovery System | Automated protein expression and purification | Enables rapid protein production (DNA to purified protein in <48 hours) for validating AI-predicted targets [31] |
| JUMP-CP Cell Painting Consortium | Publicly available morphological dataset | Provides large-scale annotated dataset for training phenotypic AI models [9] |
Visualization of AI-Driven Drug Discovery Workflows
AI-Integrated Drug Discovery Workflow
AI-Driven PDD-TDD Integration Workflow
This diagram illustrates the integrated workflow combining phenotypic and target-based discovery approaches through a central AI engine, enabling continuous data exchange and hypothesis refinement between both paradigms.
Graph Neural Network Architecture for Target Identification
Graph Neural Network for Target Identification
This architecture illustrates how graph neural networks like PDGrapher integrate diverse biological data types to identify optimal therapeutic targets through causal inference and intervention simulation.
The integration of AI and ML into both phenotypic and target-based drug discovery represents a fundamental transformation in how therapeutic candidates are identified and developed. Rather than competing approaches, PDD and TDD have become complementary streams within an integrated discovery ecosystem powered by artificial intelligence. The convergence of these methodologies is accelerating target prediction and data analysis, enabling researchers to navigate the complexity of biological systems with unprecedented precision and efficiency. As evidenced by the growing pipeline of AI-discovered candidates entering clinical trials—from Insilico Medicine's TNK inhibitor for idiopathic pulmonary fibrosis to Schrödinger's TYK2 inhibitor for autoimmune diseases—these computational approaches are delivering tangible results that promise to reshape therapeutic development [15].
Looking forward, several key trends will define the next evolution of AI in drug discovery. The merger of Recursion and Exscientia exemplifies the movement toward integrated platforms that combine massive biological data generation with sophisticated generative chemistry capabilities [15]. The emphasis on responsible AI implementation will grow, with increased focus on transparency, explainability, and bias mitigation in algorithmic decision-making [33] [31]. Additionally, the successful application of foundation models trained on extensive biological datasets will enable more accurate predictions across diverse disease contexts and patient populations [31]. As these technologies mature, we can anticipate AI-driven discovery platforms that not only accelerate the identification of therapeutic candidates but also improve clinical success rates through better target selection and patient stratification.
For researchers and drug development professionals, mastering these AI methodologies is becoming essential rather than optional. The most successful organizations will be those that effectively integrate human expertise with computational power, creating collaborative workflows that leverage the strengths of both. By embracing the integrated AI-driven approach to drug discovery outlined in this technical guide, the research community can look forward to a future where developing effective treatments for complex diseases becomes increasingly systematic, predictable, and successful.
The long-standing dichotomy in drug discovery between phenotypic drug discovery (PDD) and target-based drug discovery (TDD) is being reconciled through the integration of multi-omics technologies. Traditional TDD, which focuses on modulating specific molecular targets, often fails to capture the complexity of biological systems, contributing to high late-stage attrition rates [34]. Conversely, PDD, which observes compound effects in whole cells or organisms without presupposing targets, can identify promising compounds but often lacks mechanistic understanding [34]. Multi-omics—the integrated analysis of genomic, transcriptomic, proteomic, and other molecular datasets—provides the biological context needed to bridge this gap, offering a systems-level perspective that enhances both approaches [35].
This technical guide explores how multi-omics data informs drug discovery by elucidating complex biological mechanisms. We examine specific methodologies for multi-omics integration, showcase applications through case studies, and provide practical resources for implementing these approaches to advance both PDD and TDD pipelines.
Multi-Omics Technologies and Their Functional Contributions
Each omics layer provides unique and complementary insights into biological systems. When integrated, they enable researchers to distinguish causal disease drivers from mere associations, identifying more translatable therapeutic targets [35].
Table 1: Functional Contributions of Individual Omics Layers in Drug Discovery
| Omics Layer | Biological Information Captured | Primary Applications in Drug Discovery |
|---|---|---|
| Genomics | DNA sequence and variation, including single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) [36] | Identification of hereditary disease risk factors and patient stratification biomarkers [37] |
| Transcriptomics | RNA expression levels, revealing actively transcribed genes and alternative splicing events [34] | Understanding direct cellular responses to perturbations; biomarker discovery for treatment response [34] [35] |
| Proteomics | Protein abundance, post-translational modifications, and signaling pathways [34] | Direct profiling of therapeutic targets and understanding mechanism of action; identifying signaling network perturbations [34] [35] |
| Translatomics | Identification of RNA transcripts actively being translated into proteins [35] | Distinguishing between transcriptional and translational regulation; providing crucial intermediate layer between transcriptomics and proteomics [35] |
| Metabolomics | Dynamic levels of small-molecule metabolites and biochemical pathway activity [34] | Revealing functional outputs of cellular processes and disease-induced biochemical changes [34] |
The true power of multi-omics emerges from vertical integration across these layers. For instance, while genomics can identify disease-associated mutations, not all mutations lead to functional consequences. Layering transcriptomics, translatomics, and proteomics enables researchers to distinguish causal mutations from inconsequential ones by tracking their effects through the molecular cascade [35]. This integration is particularly valuable in PDD, where observed phenotypic changes can be "reverse engineered" to understand their molecular basis, adding mechanistic context to phenotypic hits [34].
Methodological Framework for Multi-Omics Integration
Effective multi-omics integration requires sophisticated computational approaches to handle data heterogeneity, noise, and high dimensionality. Several methodological frameworks have emerged as standards in the field.
Network-Based Integration Methods
Biological systems are inherently networked, with molecules interacting to form complex pathways and regulatory circuits. Network-based integration methods leverage this organization by representing different omics layers within unified biological networks [36].
Table 2: Categories of Network-Based Multi-Omics Integration Methods
| Method Category | Key Principles | Representative Applications |
|---|---|---|
| Network Propagation/Diffusion | Models flow of information through biological networks to identify significantly perturbed regions [36] | Prioritizing disease genes, identifying subnetworks enriched for disease mutations [36] |
| Similarity-Based Approaches | Integrates multi-omics data by computing similarity networks across patients or samples [36] | Patient stratification, drug repurposing based on similar molecular profiles [36] |
| Graph Neural Networks (GNNs) | Applies deep learning to graph-structured data for prediction and feature learning [36] | Predicting drug response, identifying novel drug-target interactions [36] |
| Network Inference Models | Reconstructs causal networks from correlation patterns in multi-omics data [36] | Elucidating regulatory relationships, identifying key drivers of disease phenotypes [36] |
These approaches are particularly valuable for contextualizing PDD findings. For example, when a compound produces a phenotypic effect in screening, network propagation can identify the subcellular networks and pathways most likely responsible, even without precise target identification [34].
AI and Machine Learning Integration
Artificial intelligence, particularly machine learning and deep learning, can detect patterns in high-dimensional multi-omics datasets that transcend human analytical capabilities [34] [38]. These models can integrate heterogeneous data types—including genomic, transcriptomic, and proteomic profiles—with phenotypic readouts to predict compound efficacy, toxicity, and mechanism of action [34] [35].
Advanced AI platforms like PhenAID exemplify this approach by integrating cell morphology data from phenotypic screening with multi-omics layers to identify patterns correlating with mechanism of action [34]. Similarly, large language models (LLMs) originally developed for natural language processing are being adapted to handle biological "languages" encoded in omics data, capturing complex patterns and inferring missing information [39].
Figure 1: Multi-Omics Data Integration Workflow for Drug Discovery
Experimental Protocols and Workflows
Integrated Multi-Omics Screening Protocol
This protocol outlines a standardized workflow for conducting multi-omics profiling following phenotypic screening to add biological context to hits.
Sample Preparation and Phenotypic Screening
- Cell Model Selection: Use physiologically relevant cell models, preferably patient-derived cells or iPSC-derived models that maintain disease-relevant phenotypes [34].
- Compound Treatment: Apply compounds at multiple concentrations (typically 3-10 concentrations for dose-response) with appropriate controls (vehicle and positive controls).
- High-Content Phenotypic Screening: Implement high-content imaging using assays like Cell Painting that visualize multiple organelle systems [34]. Fix cells at relevant timepoints (e.g., 24h, 48h, 72h) to capture phenotypic dynamics.
Multi-Omics Profiling
- Parallel Sample Processing: Split cells from the same treatment conditions for different omics analyses to ensure data comparability.
- Genomic Profiling: Extract DNA for whole-genome or exome sequencing to identify genetic variants that might influence compound response [35].
- Transcriptomic Profiling: Extract RNA for bulk or single-cell RNA sequencing. For single-cell analyses, use platform such as 10X Genomics to capture cellular heterogeneity in response to treatment [34] [35].
- Proteomic Profiling: Implement mass spectrometry-based proteomics. Utilize data-independent acquisition (DIA) methods like SWATH-MS for comprehensive protein quantification [35]. Consider phosphoproteomics to capture signaling changes.
Data Integration and Analysis
- Data Preprocessing: Normalize each omics dataset separately using appropriate methods (e.g., TPM for RNA-seq, imputation for missing values in proteomics).
- Multi-Omics Integration: Apply network-based integration methods or AI models to identify concordant and discordant patterns across omics layers [36].
- Biological Contextualization: Map multi-omics changes to phenotypic readouts to build mechanistic models of compound action [34].
Perturb-seq for Functional Genomics
Perturb-seq combines CRISPR-based gene perturbations with single-cell RNA sequencing to directly link genetic perturbations to transcriptional outcomes and phenotypic effects [34].
Experimental Workflow
- Design and Library Construction: Create a pooled CRISPR guide RNA library targeting genes of interest alongside non-targeting controls.
- Viral Transduction: Transduce cells at low MOI to ensure single perturbations.
- Single-Cell RNA Sequencing: Profile cells using 10X Genomics Chromium platform or similar.
- Perturbation Detection: Identify gRNAs from cDNA libraries during sequencing.
- Differential Expression Analysis: Compare transcriptomic profiles between targeted and control cells.
- Network Analysis: Map transcriptional changes to protein-protein interaction networks to identify affected pathways [34].
Case Studies and Applications
COVID-19 Drug Repurposing
The DeepCE model predicted gene expression changes induced by novel chemicals, enabling high-throughput phenotypic screening for COVID-19 [34]. This approach generated new lead compounds consistent with clinical evidence by integrating phenotypic and omics data with AI, demonstrating the power of multi-omics for rapid drug repurposing [34].
Cancer Target Identification
In triple-negative breast cancer, the idTRAX machine learning-based approach has been used to identify cancer-selective targets by integrating multiple omics datasets [34]. Similarly, the Archetype AI platform identified AMG900 and new invasion inhibitors using patient-derived phenotypic data integrated with omics [34].
Opioid Use Disorder (OUD) Research
Multi-omics approaches are being applied to complex neuropsychiatric conditions like OUD, where multi-omics helps unravel complex interactions between genetics, brain circuitry, immune response, and environmental stressors [38]. By combining this data with AI-driven simulations, researchers can identify new molecular targets and stratify patient populations for a condition where one-size-fits-all approaches have largely failed [38].
Research Reagent Solutions
Successful multi-omics studies require specialized reagents and platforms tailored for integrated analyses.
Table 3: Essential Research Reagents and Platforms for Multi-Omics Studies
| Reagent/Platform | Function | Application Notes |
|---|---|---|
| Cell Painting Assay Kits | Fluorescent dyes that label multiple organelles for high-content phenotypic screening [34] | Enables standardized morphological profiling; compatible with subsequent omics analyses [34] |
| Perturb-seq Libraries | Pooled CRISPR guides for large-scale genetic perturbation studies [34] | Enables linking genetic perturbations to transcriptomic and phenotypic outcomes [34] |
| Single-Cell RNA-seq Kits | reagents for capturing and barcoding single-cell transcriptomes | Critical for capturing cellular heterogeneity in response to treatments [34] [35] |
| Mass Spectrometry-Grade Proteomics Kits | Sample preparation reagents for LC-MS/MS proteomics | Enable comprehensive protein and phosphoprotein quantification [35] |
| Multi-Omics AI Platforms (e.g., PhenAID) | Software platforms integrating image-based phenotypes with omics data [34] | Provide automated analysis pipelines for mechanism of action prediction and target identification [34] |
Challenges and Future Directions
Despite its promise, multi-omics integration faces several significant barriers. Data integration remains technically complex due to heterogeneous data with varying scales, resolutions, and noise levels [35]. Infrastructure limitations represent another bottleneck, as multi-omics approaches generate enormous volumes of data requiring advanced storage and processing resources [35]. Additionally, cost considerations, regulatory concerns, and interpretability challenges of complex AI models continue to hinder widespread adoption [34] [35].
Future developments will likely focus on several key areas. First, single-cell and spatial multi-omics technologies will mature, enabling researchers to map molecular activity at the level of individual cells within their tissue context [35]. Second, AI and LLMs specifically designed for biological data will become more sophisticated, better capturing the temporal dynamics of disease processes and treatment responses [39]. Finally, efforts to standardize data formats, establish shared repositories, and develop more interpretable models will be crucial for translating multi-omics insights into clinical benefits [36] [35].
Figure 2: Multi-Omics Integration Bridges PDD and TDD Paradigms
Multi-omics technologies provide the essential biological context needed to advance both phenotypic and target-based drug discovery. By integrating genomic, transcriptomic, proteomic, and other molecular data layers, researchers can build systems-level understanding of disease mechanisms and compound actions. While computational and practical challenges remain, the continued refinement of network-based and AI-driven integration methods promises to accelerate the identification of novel therapeutic targets and biomarkers. As these technologies mature, multi-omics integration will increasingly become a cornerstone approach for developing personalized, effective treatments across diverse disease areas.
The drug discovery landscape is primarily shaped by two divergent strategies: Phenotypic Drug Discovery (PDD) and Target-Based Drug Discovery (TDD). TDD, dominant since the 1980s, employs a reductionist approach, focusing on modulating the activity of a specific, predefined molecular target with a known role in disease. In contrast, PDD is an empirical, biology-first strategy that identifies compounds based on their beneficial effects on disease phenotypes or biomarkers in realistic, often complex, biological systems, without a pre-specified target hypothesis [2]. Historically, PDD was the origin of most medicines, and after a period of focus on TDD, it has experienced a major resurgence since 2011. This revival was triggered by the observation that a majority of first-in-class drugs approved between 1999 and 2008 were discovered through phenotypic approaches [2]. Modern PDD combines the original concept with contemporary tools, systematically pursuing drug discovery based on therapeutic effects in physiologically relevant disease models. This whitepaper delves into notable case studies of successful drugs from both paradigms, providing an in-depth technical guide for researchers and drug development professionals.
Phenotypic Drug Discovery (PDD): Success Stories and Mechanisms
PDD has been notably successful in delivering first-in-class medicines with novel mechanisms of action (MoA), often expanding the "druggable" target space [2]. The following case studies exemplify this success.
Case Study: Ivacaftor (VX-770) and Elexacaftor (VX-445) for Cystic Fibrosis
- Therapeutic Context: Cystic fibrosis (CF) is a progressive genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. These mutations decrease CFTR function or disrupt its intracellular folding and trafficking to the plasma membrane [2].
- PDD Approach & Experimental Protocol: Target-agnostic compound screens were conducted using cell lines expressing wild-type or disease-associated CFTR variants (e.g., F508del, G551D). The primary readout was the restoration of CFTR channel function, measured by halide-sensitive fluorescent dyes or electrophysiology (e.g., USsing chamber assays) [2].
- Mechanism of Action: The screen identified two distinct classes of compounds:
- Potentiators (e.g., Ivacaftor): These small molecules improve the channel gating properties of CFTR mutants that reach the cell surface but have impaired opening.
- Correctors (e.g., Tezacaftor, Elexacaftor): These compounds have an unexpected MoA, enhancing the folding, stability, and plasma membrane insertion of mutated CFTR proteins [2].
- Outcome: The triple combination of elexacaftor, tezacaftor, and ivacaftor was approved in 2019 and addresses the underlying cause of CF in approximately 90% of patients [2].
The diagram below illustrates the phenotypic screening workflow for identifying CFTR modulators.
Case Study: Risdiplam for Spinal Muscular Atrophy (SMA)
- Therapeutic Context: Type 1 SMA is a rare neuromuscular disease with high infant mortality, caused by loss-of-function mutations in the SMN1 gene. A nearly identical backup gene, SMN2, exists but a splicing mutation leads to the exclusion of exon 7, producing an unstable, shorter SMN protein [2].
- PDD Approach & Experimental Protocol: Phenotypic screens were conducted using patient-derived cells or reporter cell lines where the production of full-length SMN protein from the SMN2 gene was the primary readout. High-throughput assays, such as immunoassays (ELISA) or luciferase-based splicing reporters, were employed [2].
- Mechanism of Action: The screens identified small molecules that modulate SMN2 pre-mRNA splicing. Risdiplam works by binding to two distinct sites on the SMN2 pre-mRNA, stabilizing the interaction with the U1 snRNP complex and promoting the inclusion of exon 7. This leads to increased production of full-length, functional SMN protein [2]. This represents an unprecedented drug target and MoA.
- Outcome: Risdiplam was approved by the FDA in 2020 as the first oral disease-modifying therapy for SMA [2].
Case Study: Daclatasvir for Hepatitis C Virus (HCV)
- Therapeutic Context: Hepatitis C is a liver disease caused by HCV, which infects millions globally. Treatment was revolutionized by combinations of orally available direct-acting antivirals (DAAs) [2].
- PDD Approach & Experimental Protocol: A phenotypic screen was performed using a HCV replicon system. This is a subgenomic viral RNA that replicates autonomously in human hepatoma cells, mimicking key stages of the viral lifecycle without producing infectious particles. The assay measured the inhibition of HCV RNA replication [2].
- Mechanism of Action: The screen identified compounds that potently inhibited viral replication. The molecular target was later identified as the HCV NS5A protein, a non-enzymatic protein essential for viral replication whose function was poorly characterized at the time [2].
- Outcome: Daclatasvir, an NS5A inhibitor, became a key component of DAA combinations that now cure over 90% of HCV-infected patients [2].
The Scientist's Toolkit: Key Reagents for PDD
Table 1: Essential Research Reagent Solutions for Phenotypic Drug Discovery
| Reagent / Solution | Function in PDD | Example from Case Studies |
|---|---|---|
| Disease-Relevant Cell Models | Engineered or patient-derived cells that recapitulate key aspects of the disease pathophysiology for screening. | Cell lines expressing mutant CFTR [2]; SMA patient-derived cells with SMN2 gene [2]. |
| Phenotypic Reporter Assays | Assays that quantitatively measure a disease-relevant phenotypic output, such as protein expression, localisation, or function. | Halide-sensitive fluorescent dyes for CFTR function [2]; SMN2 splicing reporter assays [2]. |
| High-Content Imaging & Analysis | Automated microscopy and image analysis to extract multiparametric data on cell morphology, protein localization, and other complex phenotypes. | Used in Cell Painting assays to profile compound effects [14]. |
| 3D Organoids / Microphysiological Systems | Advanced cell cultures that better mimic the structure and function of human tissues and organs for more physiologically relevant screening. | Increasingly used to enhance the translational relevance of PDD [8]. |
| Functional Genomic Tools (e.g., CRISPR Libraries) | Used to validate targets post-screening and deconvolute the mechanism of action of phenotypic hits. | CRISPR screening can identify genes that modulate sensitivity or resistance to phenotypic hits [40]. |
Target-Based Drug Discovery (TDD): Success Stories and Mechanisms
TDD begins with a hypothesis about the therapeutic relevance of a specific protein or gene. The following case studies illustrate the power of this approach, particularly when combined with modern technologies.
Case Study: Imatinib (Gleevec) for Chronic Myeloid Leukemia (CML)
- Therapeutic Context: CML is characterized by the Philadelphia chromosome, a genetic translocation that creates the BCR-ABL fusion gene. This gene produces a constitutively active tyrosine kinase that drives uncontrolled cell proliferation [2].
- TDD Approach & Experimental Protocol:
- Target Identification & Validation: BCR-ABL was identified as the central oncogenic driver in CML.
- Biochemical Assay Development: High-throughput screens were developed using purified BCR-ABL kinase enzyme and measured inhibition of its enzymatic activity (e.g., using ATP consumption or phosphorylation of a substrate).
- Lead Optimization: Hits from the screen were chemically optimized to improve potency, selectivity, and drug-like properties.
- Mechanism of Action: Imatinib is a potent and selective ATP-competitive inhibitor of the BCR-ABL tyrosine kinase. It binds to the inactive conformation of the kinase domain, preventing ATP binding and subsequent phosphorylation of downstream substrates, thereby halting the proliferation of CML cells [2].
- Outcome: Imatinib was approved in 2001 and revolutionized CML treatment, dramatically improving patient survival and establishing a paradigm for targeted cancer therapy.
Case Study: Modern TDD Enabled by CRISPR and AI
While Imatinib is a classic example, modern TDD is increasingly powered by functional genomics and artificial intelligence.
- CRISPR-Cas9 Screening: This technology has redefined therapeutic target identification by providing a precise and scalable platform for functional genomics [40].
- Experimental Protocol: The development of extensive single-guide RNA (sgRNA) libraries enables high-throughput screening. A typical workflow involves:
- Library Design: A pooled sgRNA library targeting thousands of genes is designed.
- Library Delivery: The library is delivered into a disease-relevant cell model (e.g., a cancer cell line) via lentiviral transduction.
- Selection Pressure: Cells are subjected to a selective pressure, such as treatment with a drug or a condition that enriches for a specific phenotype (e.g., cell survival or death).
- Next-Generation Sequencing (NGS): The abundance of each sgRNA in the population before and after selection is determined by NGS.
- Bioinformatic Analysis: Statistical analysis identifies genes whose knockout alters the cell's response to the selective pressure, revealing potential drug targets or mechanisms of resistance [40].
- Application: This approach is broadly used to identify synthetic lethal interactions in cancer, host factors for infectious diseases, and drivers of neurodegenerative diseases [40].
- Experimental Protocol: The development of extensive single-guide RNA (sgRNA) libraries enables high-throughput screening. A typical workflow involves:
The following diagram outlines the key steps in a CRISPR-Cas9 screening workflow for target identification.
- AI-Powered In Silico Screening: Artificial intelligence and machine learning are now frontline tools in TDD.
- Experimental Protocol:
- Data Curation: Large datasets of chemical structures, protein targets, and biological activities are compiled.
- Model Training: Machine learning models (e.g., deep graph networks, random forests) are trained to predict binding affinity, biological activity, or ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties.
- Virtual Screening: These models are used to computationally screen millions of compounds in silico, prioritizing a small subset for synthesis and experimental testing [41].
- Application: A 2025 study used deep graph networks to generate over 26,000 virtual analogs, leading to the discovery of sub-nanomolar inhibitors of MAGL with a 4,500-fold potency improvement over initial hits [41].
- Experimental Protocol:
The Scientist's Toolkit: Key Reagents for Modern TDD
Table 2: Essential Research Reagent Solutions for Target-Based Drug Discovery
| Reagent / Solution | Function in TDD | Example from Case Studies |
|---|---|---|
| Recombinant Proteins | Purified, often recombinant, target proteins for use in high-throughput biochemical assays. | Purified BCR-ABL kinase domain for imatinib screening [2]. |
| CRISPR sgRNA Libraries | Comprehensive pools of guide RNAs for high-throughput gene knockout studies to validate target biology and MoA. | Genome-wide libraries for identifying gene-drug interactions [40]. |
| Target Engagement Assays (e.g., CETSA) | Methods to confirm and quantify direct drug-target binding in a physiologically relevant cellular context. | CETSA used to validate engagement of DPP9 in rat tissue [41]. |
| AI/ML Software Platforms | Software for molecular property prediction, virtual screening, and de novo molecular design. | Platforms like AutoDock and SwissADME for predicting binding and drug-likeness [41]. |
| Organoid & Complex Co-cultures | Advanced cellular models for validating target relevance and compound efficacy in a tissue-like context. | Organoid-based CRISPR screening for efficient target identification [40]. |
Comparative Analysis: PDD vs. TDD
The choice between PDD and TDD is strategic and depends on the project's goals, the state of biological knowledge, and the complexity of the disease.
Table 3: Strategic Comparison of Phenotypic vs. Target-Based Drug Discovery
| Parameter | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| Starting Point | Disease phenotype in a biologically complex system. | A predefined molecular target with a hypothesized role in disease. |
| Key Strength | Discovers first-in-class drugs with novel MoAs; expands "druggable" space; suitable for polygenic diseases. | Rational, efficient, and scalable; allows for precise optimization of selectivity and potency. |
| Primary Challenge | Target deconvolution can be difficult and time-consuming; assays can be complex and low-throughput. | Requires a deep understanding of disease biology; risk of clinical failure if target hypothesis is incorrect. |
| Success Rate (First-in-Class) | Historically high for first-in-class medicines [2]. | Effective for "best-in-class" drugs following target validation by a first-in-class agent. |
| Mechanism of Action | Often uncovered after efficacy is established; can be unexpected and novel (e.g., splicing modulation). | Defined from the outset; compounds are optimized for a specific, known molecular interaction. |
| Suitability | Ideal when no attractive target is known or for diseases with complex, multifactorial etiology. | Ideal when the disease-driving target is well-validated and has a "druggable" binding site. |
A significant concept highlighted by PDD is the re-examination of polypharmacology—the ability of a single drug to interact with multiple targets. While traditionally viewed as a source of side effects, polypharmacology is increasingly recognized as a contributor to clinical efficacy, particularly in complex diseases like cancer and central nervous system disorders [2]. For example, imatinib's efficacy in multiple cancers is attributed to its inhibition of not only BCR-ABL but also c-KIT and PDGFR [2]. PDD, by being target-agnostic, is well-suited to identify such multi-target agents.
Both PDD and TDD are powerful, complementary engines for drug discovery. PDD has proven exceptionally capable of generating first-in-class drugs with novel mechanisms, as demonstrated by ivacaftor, risdiplam, and daclatasvir. TDD provides a rational path to potent and selective agents once a target is validated, exemplified by imatinib and accelerated by CRISPR and AI.
The future lies in the strategic integration of both approaches, leveraging the strengths of each. Key trends shaping this integrated future include:
- The rise of AI and Multimodal Foundation Models: Tools like PhenoModel are being developed to connect molecular structures directly with phenotypic information, accelerating the identification of bioactive compounds [14].
- Advanced Disease Models: The use of human-based microphysiological systems, organoids, and complex co-cultures will enhance the translational relevance of both PDD and TDD screens [8] [40].
- Functional Genomics: CRISPR screening will continue to be vital for target identification, MoA deconvolution, and understanding drug resistance [40].
- Emphasis on Target Engagement: Technologies like CETSA that confirm direct target binding in physiologically relevant environments are becoming standard for derisking projects [41].
For researchers, the decision to pursue a PDD or TDD strategy should be guided by the biological question at hand. PDD is the path of choice for pioneering novel biology and therapeutics, while TDD offers a powerful means to precisely engage a known pathway. The evolving toolkit, rich with functional genomics, AI, and sophisticated models, promises to enhance the success of both paradigms in delivering the next generation of medicines.
Navigating Challenges: Practical Solutions for PDD and TDD Pipelines
The PDD Landscape and the Target Deconvolution Challenge
Phenotypic Drug Discovery (PDD) has re-emerged as a powerful strategy for identifying first-in-class therapeutics, particularly for complex diseases with unmet needs. Unlike Target-Based Drug Discovery (TDD), which begins with a known molecular target, PDD identifies compounds based on their ability to modulate a disease-relevant phenotype in a biologically complex system, without a preconceived target hypothesis [2]. This empirical, biology-first approach has led to a disproportionate number of innovative medicines but introduces a central challenge: the subsequent identification of the compound's molecular mechanism of action (MoA), a process known as target deconvolution [42] [2].
Successfully elucidating the MoA is not merely an academic exercise; it is critical for lead optimization, safety profiling, and designing clinical biomarkers. The challenge is pronounced because phenotypic hits often act through novel or unexpected targets, including multi-component cellular machines, or exhibit polypharmacology (engagement of multiple targets) that collectively underpin the efficacy [2]. This guide details the advanced strategies and methodologies enabling researchers to overcome this major hurdle, thereby accelerating the development of novel therapeutics from phenotypic screens.
Table 1: Notable Drugs Discovered Through PDD and Their Deconvoluted Mechanisms
| Drug Name | Indication | Phenotypic Screen | Deconvoluted Mechanism of Action (MoA) |
|---|---|---|---|
| Daclatasvir | Hepatitis C | HCV replicon assay [2] | Potent modulator of the HCV NS5A protein, an essential viral protein with no known enzymatic function [2]. |
| Ivacaftor, Tezacaftor, Elexacaftor | Cystic Fibrosis | Cell lines expressing mutant CFTR [2] | Ivacaftor is a "potentiator" that improves CFTR channel gating; correctors (elexacaftor, tezacaftor) enhance CFTR folding and trafficking [2]. |
| Lenalidomide | Multiple Myeloma | Observations of efficacy in leprosy and multiple myeloma [2] | Binds to the E3 ubiquitin ligase Cereblon, altering its substrate specificity to promote degradation of transcription factors IKZF1/IKZF3 [2] [43]. |
| Risdiplam | Spinal Muscular Atrophy | SMN2 splicing modifiers [2] | Modulates SMN2 pre-mRNA splicing by stabilizing the U1 snRNP complex, increasing full-length SMN protein [2]. |
Strategic Frameworks for MoA Elucidation
Modern MoA elucidation is a multi-faceted process that integrates several technological approaches. Rather than relying on a single method, a convergent evidence strategy is employed, where data from complementary techniques are combined to build a compelling hypothesis for the compound's biological activity.
The Integrated Workflow for Target Deconvolution
A systematic approach begins with profiling the phenotypic hit to generate hypotheses, which are then validated through direct target engagement assays. The following diagram illustrates this integrated workflow.
Hypothesis Generation through Pathway Profiling
The first step is to generate plausible hypotheses about the pathways and processes a compound affects.
- High-Content Phenotypic and Pathway Profiling: This involves using high-content imaging and transcriptomics or proteomics to create a detailed fingerprint of the compound's effect on the cell. Machine learning algorithms then compare this fingerprint to those of compounds with known MoAs in large databases. A high similarity score can suggest a shared target or pathway, rapidly providing a testable MoA hypothesis [42].
- Functional Genomics Screening: Techniques like genome-wide CRISPR-Cas9 knockout screens are used to identify genes whose loss either sensitizes cells to or protects them from the compound's effect. If knocking out a specific gene confers resistance, the product of that gene is a strong candidate for being the compound's direct target or a critical component in its pathway [2].
- Chemical Profiling: Assessing the compound's chemical similarity to known tool compounds can provide initial, albeit low-resolution, clues about its potential target class.
Hypothesis Validation through Direct Target Engagement
After generating hypotheses, the focus shifts to directly identifying the physical target(s).
- Affinity-Based Pulldown and Proteomics: This is a direct biochemical approach. The phenotypic hit is chemically modified with an affinity tag (e.g., biotin) without destroying its bioactivity. This "bait" molecule is then incubated with a cell lysate or introduced into live cells to bind its protein targets. The bait-protein complex is purified using beads coated with streptavidin, and the co-purifying proteins are identified via mass spectrometry. This method was pivotal in identifying Cereblon as the target of lenalidomide [43].
- Cellular Target Engagement Assays: Techniques like Cellular Thermal Shift Assay (CETSA) and its variants monitor drug-target interactions in a native cellular environment. When a small molecule binds to a protein, it often changes the protein's thermal stability. CETSA detects this shift by heating cells treated with the compound and measuring the amount of soluble target protein that remains. This confirms engagement in live cells and can be used to screen candidate targets [2].
- Functional Rescue Experiments: This genetic approach involves overexpressing a putative target protein in a cell. If the compound's phenotypic effect is diminished or abolished because the excess "sinks" out the compound, it provides strong functional evidence that the overexpressed protein is the relevant target.
Detailed Experimental Protocols
This section provides detailed methodologies for key experiments cited in this guide.
Affinity-Based Pull-down and Mass Spectrometry
This protocol is used to isolate and identify direct protein targets of a small molecule from a complex cellular lysate [43].
Key Research Reagents: Table 2: Essential Reagents for Affinity Pull-down
| Reagent | Function/Description |
|---|---|
| Biotin- or Alkyne-Tagged Analog | A functionalized, bioactive version of the hit compound that serves as the "bait". |
| Streptavidin/Solid Support | Magnetic streptavidin beads or agarose resin for immobilization and purification. |
| Cell Lysate | Source of potential protein targets, prepared from relevant cell lines. |
| Mass Spectrometry (LC-MS/MS) | For high-sensitivity identification of purified proteins. |
| Competitor (Untagged Compound) | Untreated hit compound used in competition control to confirm binding specificity. |
Procedure:
- Compound Design & Validation: Synthesize a biotin- or alkyne-tagged analog of the phenotypic hit. Critically, validate that this tagged analog retains biological activity in the original phenotypic assay.
- Lysate Preparation: Lyse relevant cells (e.g., disease model cell lines) in a non-denaturing lysis buffer to preserve protein structures and interactions. Pre-clear the lysate with bare beads to reduce non-specific binding.
- Pull-down Experiment: Incubate the cell lysate with the immobilized bait compound. In parallel, run a competition control: pre-incubate lysate with a large excess of untagged compound before adding the bait. This should outcompete and block specific binding to the bait.
- Wash and Elution: Thoroughly wash the beads with lysis buffer to remove non-specifically bound proteins. Elute bound proteins using a denaturing buffer (e.g., SDS-PAGE sample buffer) or by boiling.
- Protein Identification: Separate eluted proteins by SDS-PAGE, perform in-gel tryptic digestion, and analyze the resulting peptides by Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS). Proteins significantly enriched in the bait sample compared to the competition control are high-confidence specific binders.
High-Content Pathway Profiling
This protocol uses high-content imaging and machine learning to infer MoA based on morphological and pathway signatures [42].
Procedure:
- Cell Staining and Imaging: Seed cells in multi-well plates and treat with the phenotypic hit, reference compounds with known MoAs, and vehicle controls. After treatment, fix and stain cells with fluorescent dyes or antibodies for key cellular features (e.g., DNA, cytoskeleton, specific phospho-proteins, organelles).
- Image Feature Extraction: Acquire high-resolution images on an automated microscope. Use image analysis software to extract hundreds of quantitative morphological features (e.g., cell size, shape, texture, intensity, and organelle distribution) for each cell.
- Data Analysis and Pattern Matching: Use machine learning (e.g., principal component analysis, clustering) to analyze the multi-parametric feature data. The resulting "phenotypic fingerprint" of the unknown compound is computationally compared to the fingerprints of the reference compound library. A close match to a known MoA class provides a testable hypothesis for the hit's mechanism.
Table 3: Technology Platforms for Advanced Target Deconvolution
| Technology Platform | Primary Application in Deconvolution | Key Output |
|---|---|---|
| High-Content Imaging & Analysis | Phenotypic profiling & hypothesis generation [42] | Multiparametric cellular fingerprint for pattern matching. |
| Next-Generation Sequencing (NGS) | Functional genomics (CRISPR screens) [2] | List of genes that modulate compound sensitivity. |
| High-Resolution Mass Spectrometry | Proteomics (pulldown, phosphoproteomics) [43] | Identity of binding proteins or altered signaling pathways. |
| Cellular Thermal Shift Assay (CETSA) | Target engagement in live cells [2] | Confirmation of physical drug-target interaction. |
| Structural Biology (Cryo-EM/X-ray) | MoA confirmation & rational design [2] | Atomic-resolution structure of drug-target complex. |
Case Studies in Successful MoA Elucidation
E3 Ligase Modulators (IMiDs)
The discovery of the MoA for thalidomide and its analogs (lenalidomide, pomalidomide) is a landmark case in PDD. These IMiD drugs were developed based on clinical observations of their efficacy, but their molecular targets remained unknown for decades. The breakthrough came from an affinity-based pull-down approach, where a thalidomide derivative was used as bait to isolate Cereblon (CRBN) from cell lysates [2] [43]. Subsequent functional and biochemical studies revealed the unprecedented MoA: the drugs act as "molecular glues" that reprogram the CRL4CRBN E3 ubiquitin ligase to target novel proteins, including the transcription factors IKZF1 and IKZF3, for degradation [43]. This deconvolution opened the new field of targeted protein degradation.
Splicing Modulator for Spinal Muscular Atrophy (SMA)
SMA is caused by loss-of-function of the SMN1 gene. Phenotypic screens were designed to identify small molecules that could increase levels of functional SMN protein from the backup SMN2 gene, which is normally mis-spliced. The hit compound, later named risdiplam, emerged from such a screen. Its MoA was deconvoluted through a combination of chemical biology and biochemical studies, which revealed that it binds to two specific sites on the SMN2 pre-mRNA. By stabilizing the interaction between the mRNA and the U1 snRNP complex, it promotes the correct inclusion of exon 7, producing a stable, functional SMN protein [2]. This represented a novel MoA targeting RNA splicing.
The Scientist's Toolkit: Key Research Reagent Solutions
A successful target deconvolution campaign relies on a suite of specialized reagents and tools.
Table 4: Essential Research Reagents for Target Deconvolution
| Research Reagent / Tool | Brief Function/Explanation |
|---|---|
| Bioactive, Tagged Compound Analog | A chemically modified version of the hit (e.g., with biotin or a photo-affinity tag) used as "bait" in pull-down experiments to isolate target proteins [43]. |
| CRISPR Knockout Library | A pooled collection of guide RNAs targeting all genes in the genome, used in functional genomics screens to identify genes critical for a compound's activity [2]. |
| Phospho-Specific Antibodies | Antibodies that detect specific phosphorylated proteins; used in high-content profiling to map the signaling pathways affected by compound treatment [42]. |
| Stable Isotope Labeling (SILAC) | A mass spectrometry-based method that uses heavy and light amino acids to quantitatively compare protein levels or phosphorylation states between treated and untreated samples. |
| Mechanistic Cell-Based Models | Disease-relevant cellular models (e.g., primary cells, co-cultures, 3D organoids) that provide a biologically context for both the initial phenotypic screen and subsequent MoA studies [8]. |
The major hurdle of target deconvolution in PDD is being systematically overcome by a powerful and integrated toolkit of technologies. The convergence of high-content profiling, functional genomics, and chemical proteomics provides a multi-angled strategy to generate and validate MoA hypotheses with greater speed and confidence than ever before. As these technologies mature—particularly with the integration of more physiologically relevant human-based models and advanced machine learning for data integration—they will transform target deconvolution from a daunting bottleneck into a manageable, predictive process [42] [8]. This progress solidifies PDD's role as an indispensable engine for discovering first-in-class drugs against complex and challenging diseases.
The high failure rate of late-stage clinical trials represents one of the most significant challenges in pharmaceutical development. Approximately 90% of clinical drug development fails, with inadequate efficacy representing the primary cause of Phase III trial failures [44] [45]. This attrition problem is particularly pronounced in Target-Based Drug Discovery (TDD), a reductionist approach that focuses on modulating specific molecular targets of interest. Between 2025 and 2029, an estimated $350 billion of revenue is at risk from patent expirations, increasing pressure on R&D productivity [46]. The internal rate of return for biopharma R&D investment has fallen to just 4.1% – well below the cost of capital [46]. This perspective examines the systemic causes of efficacy failure in TDD and outlines integrated strategies to improve translational success, framed within the comparative advantages of Phenotypic Drug Discovery (PDD) approaches.
Quantitative Analysis of the Efficacy Challenge
The efficiency of pharmaceutical R&D has declined despite advances in target validation technologies. Understanding the magnitude and financial impact of this problem is crucial for implementing effective solutions.
Table 1: Clinical Trial Failure Rates and Contributing Factors
| Development Phase | Success Rate | Primary Failure Cause | Percentage of Failures | Average Cost per Trial |
|---|---|---|---|---|
| Phase I to Approval | 13.8% [45] | Overall Attrition | 86.2% [45] | $2.3B (total cost-to-market) [47] |
| Phase III | 46% (Success) [45] | Inadequate Efficacy | 57% [45] | ~$60M (Oncology) [47] |
| Phase III | 46% (Success) [45] | Safety Concerns | 17% [45] | ~$60M (Oncology) [47] |
Table 2: Impact of Nonadherence on Clinical Trial Outcomes
| Nonadherence Level | Power Reduction (from 90%) | Power Reduction (from 80%) | Required Sample Size Increase |
|---|---|---|---|
| 10% | 84% | 72% | 23% |
| 20% | 74% | 61% | 56% |
| 30% | 64% | 52% | 100% |
| 50% (Actual rate in all phases) [45] | 45% | 36% | 300% |
The crisis extends beyond failure rates to encompass unsustainable cost structures. Trials have become increasingly complex and expensive, with per-trial costs rising by 4.9% annually for multinational studies [47]. Each day of delayed drug launch costs sponsors an average of $500,000 in lost drug revenue, a figure that can exceed $3 million per day for blockbuster drugs [47]. These financial pressures, combined with the 6.7% success rate for Phase I drugs in 2024 (down from 10% a decade ago), create an urgent need for reform [46].
Root Causes of Efficacy Failure in TDD
Inadequate Target Validation
A fundamental weakness in conventional TDD pipelines lies in the transition from biochemical assays to clinical efficacy. While TDD excels at developing compounds with high potency and specificity against purified targets, this approach often overlooks tissue exposure and selectivity in disease-relevant environments [44]. The disconnect between biochemical potency and physiological relevance means that many compounds fail when they encounter the complexity of human biology.
The limitations of standard validation methods have prompted innovation in target engagement technologies. Cellular Thermal Shift Assay (CETSA) has emerged as a leading approach for validating direct target binding in intact cells and native tissue environments [41]. Recent applications have demonstrated its utility in quantifying drug-target engagement ex vivo and in vivo, providing critical data on whether compounds actually engage their intended targets in physiologically relevant systems [41].
Insufficient Attention to Tissue Exposure and Selectivity
The Structure–Tissue exposure/selectivity–Activity Relationship (STAR) framework provides a valuable classification system for understanding how tissue distribution profiles impact clinical outcomes [44]. This model categorizes drug candidates into four distinct classes:
Table 3: STAR Classification of Drug Candidates and Clinical Outcomes
| STAR Class | Specificity/Potency | Tissue Exposure/Selectivity | Clinical Dose | Efficacy/Toxicity Balance |
|---|---|---|---|---|
| Class I | High | High | Low | Superior efficacy/safety with high success rate [44] |
| Class II | High | Low | High | High toxicity with adequate efficacy; requires cautious evaluation [44] |
| Class III | Relatively low (adequate) | High | Low | Manageable toxicity with adequate efficacy; often overlooked [44] |
| Class IV | Low | Low | Variable | Inadequate efficacy/safety; should be terminated early [44] |
Traditional Structure-Activity Relationship (SAR) focused optimization frequently produces Class II drugs with high specificity but poor tissue selectivity, requiring high doses that lead to toxicity issues [44]. The STAR framework explains why many TDD-derived candidates fail in clinical development – they may show excellent biochemical potency but lack the tissue-level selectivity needed for therapeutic utility.
Clinical Trial Execution Challenges
Beyond compound optimization issues, clinical trial methodology itself contributes significantly to efficacy failure. Patient nonadherence represents a major confounder, with approximately 50% of participants across all clinical trial phases admitting to not following the dosing regimen [45]. This nonadherence can lead to underestimation of drug efficacy, overestimation of dosing requirements, and raised safety concerns – all key reasons for delay and denial of regulatory approval [45].
The problem is compounded by recruitment challenges and unrepresentative patient populations. Only 5%-8% of potentially eligible patients ever participate in clinical trials, and recruitment is heavily concentrated in urban areas near academic medical centers [47]. This creates significant barriers for patients in rural communities and limits the generalizability of trial results.
Integrated Strategies for Mitigating Efficacy Failure
Strengthening Target Validation Through Physiologically Relevant Models
Improving the predictive validity of early discovery assays requires a shift toward more complex biological systems. Phenotypic Drug Discovery (PDD) has re-emerged as a powerful alternative approach, accounting for a disproportionate number of first-in-class medicines [2]. Modern PDD combines the original concept of observing therapeutic effects on disease physiology with advanced tools and strategies, systematically pursuing drug discovery based on therapeutic effects in realistic disease models [2].
Successful applications of PDD include:
- Cystic fibrosis: Target-agnostic compound screens identified correctors that enhance CFTR folding and plasma membrane insertion through unexpected mechanisms [2]
- Spinal muscular atrophy: Phenotypic screens identified small molecules that modulate SMN2 pre-mRNA splicing via an unprecedented drug target and mechanism of action [2]
- HCV treatment: Phenotypic screening revealed the importance of NS5A, which has no known enzymatic activity, as a key component of direct-acting antiviral combinations [2]
Implementing the STAR Framework in Candidate Selection
Adopting the Structure–Tissue exposure/selectivity–Activity Relationship (STAR) framework enables more informed candidate selection and clinical dose planning [44]. This approach requires:
Experimental Protocol: Comprehensive Tissue Distribution Study
- Dosing Regimen: Administer candidate compounds to disease-relevant animal models at three dose levels (low, medium, high) for 7-14 days
- Sample Collection: Collect plasma and tissue samples (liver, kidney, target tissue, brain) at multiple time points (0.5, 1, 2, 4, 8, 12, 24 hours post-dose)
- Bioanalysis: Use LC-MS/MS to quantify compound concentrations in all matrices
- Data Analysis: Calculate AUC(0-t), Cmax, Tmax, and tissue-to-plasma ratios for each tissue
- Correlation: Establish exposure-response and exposure-toxicity relationships
This systematic evaluation of tissue distribution enables researchers to classify compounds according to the STAR framework and prioritize those with balanced specificity and tissue exposure profiles (Class I and III) [44].
Enhancing Clinical Trial Quality through Digital Adherence Monitoring
Addressing the medication nonadherence problem requires moving beyond traditional measurement methods. Digital adherence monitoring utilizing connected packaging and powerful data analytics provides objective, qualitative data that can guide informed decisions and interventions [45].
Experimental Protocol: Implementing Digital Adherence Monitoring
- Technology Selection: Deploy smart packaging solutions (connected blister packs, pre-filled syringes) with embedded sensors
- Data Collection: Capture essential parameters including time/date of dose administration, completion status, and device interaction patterns
- Platform Integration: Transmit data to cloud-based analytics platforms for sophisticated analysis of medication-taking behaviors
- Risk Stratification: Use visualization tools to identify "at-risk" participants based on dosing patterns
- Intervention: Implement personalized support strategies for non-adherent participants
Studies demonstrate that smart package monitoring is 97% accurate, compared to 60% for pill counting and just 27% for self-reporting [45]. This approach provides a complete understanding of patient adherence behaviors, enabling sponsors to distinguish true efficacy failures from adherence-related artifacts.
Leveraging Artificial Intelligence for Trial Optimization
AI-driven models are transforming clinical trial design through sophisticated simulation capabilities. Quantitative systems pharmacology (QSP) models and "virtual patient" platforms simulate thousands of individual disease trajectories, allowing researchers to test dosing regimens and refine inclusion criteria before a single patient is dosed [37].
Experimental Protocol: AI-Enhanced Trial Simulation
- Model Development: Create computational models incorporating disease pathophysiology, drug mechanism, and population variability
- Virtual Patients: Generate simulated patient populations with relevant demographic, genetic, and clinical characteristics
- Trial Simulation: Run thousands of simulated trials with varying designs, endpoints, and inclusion criteria
- Optimization: Identify optimal trial parameters that maximize statistical power while minimizing sample size and duration
- Validation: Compare simulation predictions with historical trial data to refine model accuracy
Companies like Unlearn.ai have validated digital twin-based control arms in Alzheimer's trials, demonstrating that AI-augmented virtual cohorts can reduce placebo group sizes while maintaining statistical power [37].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successfully implementing these strategies requires access to specialized research tools and technologies. The following table outlines key solutions for addressing efficacy challenges:
Table 4: Research Reagent Solutions for Efficacy Optimization
| Technology/Reagent | Function | Application in Efficacy Optimization |
|---|---|---|
| CETSA (Cellular Thermal Shift Assay) | Measures target engagement in intact cells and tissues [41] | Confirms compound binding to intended target in physiologically relevant environments [41] |
| Digital Adherence Monitoring | Electronically records medication-taking events [45] | Provides accurate adherence data to distinguish true efficacy failures from nonadherence [45] |
| Microphysiological Systems (Organ-on-a-Chip) | Models human tissue and disease environments in vitro [8] | Evaluates compound efficacy and toxicity in human-relevant systems before clinical trials [8] |
| AI-Powered Trial Simulation Platforms | Creates digital twins and virtual patient populations [37] | Optimizes trial design and predicts outcomes before patient enrollment [37] |
| Quantitative Proteomics Platforms | Measures protein expression and modification in tissues [41] | Evaluates target modulation and pathway engagement in disease-relevant tissues [41] |
Addressing the persistent challenge of efficacy failure in clinical trials requires fundamental changes in how we approach drug discovery and development. The integration of Phenotypic Drug Discovery principles into target validation, adoption of the STAR framework for candidate selection, implementation of digital adherence technologies in clinical trials, and application of AI-driven trial simulations collectively offer a path toward more predictable and successful development outcomes.
The examples of successful PDD-derived therapeutics demonstrate how target-agnostic approaches can expand the "druggable target space" to include unexpected cellular processes and novel mechanisms of action [2]. These successes, combined with modern tools for evaluating tissue exposure and engagement in physiologically relevant systems, provide a roadmap for mitigating the primary causes of efficacy failure.
As the industry faces unprecedented patent cliffs and economic pressures, embracing these integrated approaches becomes essential for sustaining innovation. By learning from both TDD and PDD paradigms and leveraging advanced technologies throughout the development process, researchers can increase the likelihood that investments in early discovery will translate to meaningful clinical benefits for patients.
The strategic dichotomy between phenotypic drug discovery (PDD) and target-based drug discovery (TDD) represents a fundamental framework in biomedical research. TDD employs a hypothesis-driven approach, focusing on modulating a specific, known molecular target, which requires a deep prior understanding of the disease's molecular underpinnings [48]. In contrast, PDD uses a more holistic, empirical strategy, screening for compounds that produce a desired observable change in cells, tissues, or whole organisms without requiring prior knowledge of the specific molecular mechanism of action (MMOA) [49] [48]. Notably, a seminal analysis revealed that phenotypic strategies have been the more successful route for discovering first-in-class small molecule medicines, largely because they allow for the unbiased identification of the MMOA [49] [10].
The modern resurgence of PDD is powered by advanced multi-modal assays that simultaneously capture diverse molecular and functional readouts from biological systems [8] [10]. These assays can profile the transcriptome, chromatin accessibility, proteome, and other molecular properties, often at a single-cell resolution [50] [51]. The integration of these complementary data types provides a holistic perspective of biological systems, offering unprecedented potential to uncover novel disease mechanisms, identify molecular subtypes, and discover new drug targets and biomarkers [52] [53]. However, this promise is contingent on successfully navigating significant data challenges, including high-dimensionality, heterogeneity, and sparsity [52]. Effectively managing these challenges is crucial for leveraging the full power of PDD in identifying novel therapeutic mechanisms that might be missed by conventional target-based approaches [48].
Core Data Challenges in Multi-Modal Assays
The integration of data from multi-modal assays is fraught with computational and analytical hurdles that stem from the intrinsic nature of the technologies and biological systems themselves. Three primary challenges dominate this landscape.
Data Heterogeneity
Data heterogeneity arises from the use of diverse laboratory techniques and technologies to generate different types of molecular data. Multi-omics datasets typically comprise thousands of features and are generated through disparate protocols, leading to inconsistent data distributions, varying scales, and distinct technical biases or batch effects across modalities [52]. For instance, single-cell RNA-sequencing (scRNA-seq) data is discrete count-based, single-cell ATAC-sequencing (scATAC-seq) data is binary, and protein abundance data from tagged antibodies often includes a nonzero background component [50]. This fundamental heterogeneity makes direct integration and comparison non-trivial, requiring sophisticated methods that can account for the unique statistical properties of each data type [50].
Data Sparsity and Missing Values
Sparsity is a ubiquitous issue in multi-omics datasets, manifesting in two key forms. First, due to experimental limitations, data quality issues, or incomplete sampling, these datasets are often unbalanced and incomplete, with missing values for specific modalities across a set of samples [52]. Second, in the context of single-cell technologies, "dropout" events occur where a gene is expressed but not detected, leading to zero-inflated data matrices [50]. This sparsity complicates the analysis, as the absence of a signal does not necessarily equate to a true biological negative. The frequency of missing values and the inherent noisiness of the data can obscure biological signals and hinder the ability to draw robust conclusions from integrated analyses.
High-Dimensionality
Multi-omics datasets are characteristically high-dimensional, often encompassing thousands of features (e.g., genes, chromatin regions, proteins) across a relatively smaller number of samples or cells [52] [51]. This "curse of dimensionality" poses a significant challenge, as traditional statistical methods may struggle with such data spaces, increasing the risk of overfitting and computational intractability. Dimensionality reduction is thus not merely a preprocessing step but a necessity to condense datasets into fewer, meaningful factors that reveal important biological patterns for downstream tasks like clustering, classification, and biomarker identification [52] [51].
Computational Integration Strategies and Methodologies
A range of computational methods has been developed to address the challenges of multi-modal data integration. These can be broadly categorized by their underlying algorithmic approach, each with distinct strengths and ideal applications.
Table 1: Overview of Multi-Omics Data Integration Methods
| Model Approach | Strengths | Limitations | Typical Applications |
|---|---|---|---|
| Correlation / Covariance-based (e.g., CCA, PLS) | Captures linear relationships, interpretable, flexible sparse extensions [52]. | Limited to linear associations, typically requires matched samples [52]. | Disease subtyping, detection of co-regulated modules [52]. |
| Matrix Factorisation (e.g., JIVE, NMF) | Efficient dimensionality reduction, identifies shared and omic-specific factors, scalable [52]. | Assumes linearity, does not explicitly model uncertainty [52]. | Disease subtyping, biomarker discovery [52]. |
| Probabilistic-based (e.g., iCluster) | Captures uncertainty in latent factors, probabilistic inference [52]. | Computationally intensive, may require strong model assumptions [52]. | Disease subtyping, latent factor discovery [52]. |
| Network-based | Robust to missing data, represents complex relationships [52]. | Sensitive to similarity metrics, may require extensive tuning [52]. | Patient similarity analysis, regulatory mechanism identification [52]. |
| Deep Generative Learning (e.g., VAEs, GANs) | Learns complex nonlinear patterns, supports missing data and denoising, flexible architectures [52] [50] [51]. | High computational demands, limited interpretability, requires large datasets [52]. | High-dimensional integration, data imputation, disease subtyping [52]. |
Classical Statistical and Machine-Learning Approaches
Classical methods provide a foundation for multi-omics integration. Canonical Correlation Analysis (CCA) and its extensions, such as sparse Generalized CCA (sGCCA), are designed to find linear combinations of variables from two or more datasets that are maximally correlated [52]. Supervised versions like DIABLO extend this framework to simultaneously maximize common information between omics datasets and minimize the prediction error of a phenotypic response variable, effectively selecting co-varying modules that explain an outcome [52].
Matrix factorization techniques, including Joint and Individual Variation Explained (JIVE) and integrative Non-Negative Matrix Factorization (intNMF), decompose multiple omics datasets into joint and individual low-rank approximations, separating shared patterns from dataset-specific noise [52]. Probabilistic methods like iCluster use a joint latent variable model to identify shared latent factors (e.g., cancer subtypes) from multi-omics data, incorporating uncertainty estimates [52].
Deep Learning-Based Integration
Deep learning models have gained prominence for their ability to identify complex, nonlinear patterns in large, heterogeneous datasets [51].
Non-generative models, such as standard autoencoders (AEs), learn a compressed, low-dimensional representation (latent space) of the input data. These are powerful for dimensionality reduction and integration but do not model the underlying data distribution, limiting their ability to generate new data or handle significant missingness [51].
Generative models, particularly Variational Autoencoders (VAEs), have become a cornerstone for modern multi-omics integration. VAEs learn the probability distribution of the data, allowing them to generate new data points and provide calibrated uncertainty estimates for their predictions [52] [50]. A key advantage is their ability to handle missing modalities and perform data imputation.
Experimental Protocol: Multi-Modal Integration with MultiVI
MultiVI is a deep probabilistic model that exemplifies the application of VAEs for integrating single-cell multi-omics data, such as scRNA-seq and scATAC-seq [50]. The detailed methodology is as follows:
- Input: A multimodal dataset ( X ) is divided into modality-specific observations (e.g., ( XR ) for gene expression, ( XA ) for chromatin accessibility), along with sample batch information ( S ).
- Encoding: Two separate deep neural network encoders learn to map each modality to a batch-corrected, multivariate normal distribution in a latent space: ( q(zR | XR, S) ) and ( q(zA | XA, S) ).
- Integration: The model minimizes the distance between the two latent representations. The final, integrative cell state ( q(z | XR, XA, S) ) is computed as the average of the two modality-specific representations.
- Handling Missing Modalities: For cells where only one modality is available (unpaired), the latent state is drawn directly from the available modality's encoder.
- Decoding and Generation: Modality-specific decoder networks reconstruct the observed data from the joint latent representation ( z ). RNA expression is modeled with a Negative Binomial distribution, and accessibility data with a Bernoulli distribution, accounting for their distinct statistical properties.
- Adversarial Component: An adversarial training step is included to penalize the model if cells from different modalities become overly separated in the latent space, ensuring effective integration [50].
This architecture allows MultiVI to create a joint representation that facilitates the analysis of all modalities, even for cells where one or more modalities are missing, enabling tasks like clustering, visualization, and the imputation of missing data.
The Scientist's Toolkit: Essential Research Reagents and Platforms
The successful execution of multi-modal assays and the subsequent validation of findings rely on a suite of critical research reagents and technological platforms.
Table 2: Key Research Reagent Solutions for Multi-Modal Assays
| Reagent / Platform | Function | Application in PDD & TDD |
|---|---|---|
| High-Content Imaging Systems | Automated, high-resolution microscopy for quantifying complex phenotypic changes in cells (e.g., morphology, protein localization) [8]. | Core to PDD for unbiased assessment of compound effects in disease-relevant cell models. |
| CRISPR Screening Tools | Enables genome-wide or targeted gene knockout/modulation to identify genes essential for a phenotype or compound sensitivity [10]. | Used in both PDD (for target deconvolution) and TDD (for target validation). |
| Patient-Derived Cell Models | Primary cells or organoids sourced from patients with the disease, preserving key pathological features [10]. | Crucial for PDD to ensure biological relevance and improve translational predictability. |
| Multimodal Single-Cell Protocols (e.g., 10x Multiome) | Technologies for concomitantly profiling gene expression, chromatin accessibility, and protein abundance in the same single cell [50] [53]. | Provides a unified view of cellular states, informing both PDD and TDD. |
| Multimodal Nanosensors | Probes for real-time monitoring of conditions within the tumor microenvironment (TME) or other complex biological niches [53]. | Enables dynamic, functional readouts in complex systems relevant to PDD. |
Application in Drug Discovery: Connecting Data to Therapeutics
The integration of multi-modal data is transforming both phenotypic and target-based drug discovery paradigms. In PDD, these approaches are instrumental in target deconvolution—the process of identifying the molecular mechanism of action (MMOA) of a compound identified in a phenotypic screen [49] [8]. By analyzing multi-omics profiles of cells treated with a hit compound, researchers can use integration methods to infer which pathways or targets are modulated, thereby converting a phenotypic hit into a target hypothesis.
Furthermore, multi-modal integration enhances tumor microenvironment (TME) characterization in oncology. The TME plays a crucial role in tumor progression and therapy response. Advances in single-cell and spatial multi-omics technologies provide fine-grained resolution of the TME [53]. For example, integrating single-cell RNA-seq with multiplexed ion beam imaging has identified distinct tumor subgroups and rare cell types, revealing cellular interactions that underlie resistance to immunotherapy [53]. This comprehensive characterization is vital for developing new cancer therapies, whether through phenotypic screens for compounds that alter the TME or target-based approaches against newly discovered interactions.
In the context of complex diseases like Alzheimer's, where the exact molecular mechanisms are not fully understood, phenotypic screening combined with multi-omics profiling offers a pathway to uncover novel therapeutic mechanisms that might be missed by conventional target-based approaches focused on single molecules like amyloid-beta [48]. The holistic view provided by multi-modal data integration is thus critical for supporting the future of phenotypic drug discovery.
The drug discovery landscape has long been characterized by two dominant but often competing strategies: Phenotypic Drug Discovery (PDD) and Target-Based Drug Discovery (TDD). Historically, PDD involved identifying compounds that modify disease phenotypes without prior knowledge of specific molecular targets, allowing for the discovery of drugs acting through previously unknown mechanisms [54]. In contrast, TDD aims to find drugs that interact with a specific, pre-validated target molecule believed to play a crucial role in the disease process, offering advantages in specificity and reduced off-target effects [54]. Between 1999 and 2008, a surprising observation emerged that a majority of first-in-class drugs were discovered empirically without a drug target hypothesis, leading to a major resurgence of PDD since 2011 [2]. However, in recent years, researchers have increasingly recognized the value of integrating these approaches in a complementary manner, creating innovative hybrid strategies that combine their strengths for more effective drug development processes [54].
This evolution toward hybrid models represents a paradigm shift in pharmaceutical research, moving beyond the traditional dichotomy to embrace a more holistic and pragmatic approach. By leveraging the target-agnostic, biology-first strength of PDD while incorporating the precision and mechanistic understanding of TDD, these integrated approaches offer unprecedented opportunities to address previously intractable challenges in drug discovery. This whitepaper explores the scientific rationale, methodological frameworks, and practical implementation of these hybrid approaches, providing researchers with both theoretical foundations and practical protocols for their application in modern drug development pipelines.
Comparative Analysis: PDD vs. TDD Strengths and Limitations
A comprehensive understanding of both PDD and TDD approaches is essential for effectively integrating them into a cohesive strategy. The following table summarizes the core characteristics, advantages, and limitations of each approach:
Table 1: Comparative Analysis of PDD and TDD Approaches
| Aspect | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| Core Principle | Identification of compounds that modify disease phenotype without prior target knowledge [54] | Finding drugs that interact with a specific, pre-validated molecular target [54] |
| Key Advantages | - Discovers novel mechanisms and targets- Expands "druggable" target space- Identifies polypharmacology opportunities [2] | - Increased specificity- Reduced off-target effects- More straightforward optimization [54] |
| Major Limitations | - Complex target deconvolution- Risk of advancing undesirable mechanisms- Challenges in hit triage and prioritization [8] | - Limited to known biology- May miss complex biological interactions- Restricted to traditionally "druggable" targets [54] |
| Success Examples | - Ivacaftor/lumacaftor (cystic fibrosis)- Risdiplam (spinal muscular atrophy)- Lenalidomide (multiple myeloma) [2] | - Imatinib (CML)- Most kinase inhibitors [2] |
| Target Identification | Required after compound identification (often challenging) [8] | Defined before compound screening (straightforward) |
| Therapeutic Areas | Particularly strong for complex, polygenic diseases [2] | Effective for well-characterized molecular pathways |
The disproportionate number of first-in-class medicines derived from PDD approaches highlights its value in expanding "druggable" target space [2]. Notable examples include daclatasvir (HCV NS5A modulator), cystic fibrosis correctors (elexacaftor, tezacaftor), and risdiplam (SMN2 splicing modifier), all of which emerged from phenotypic screens and revealed unprecedented mechanisms of action [2]. Conversely, TDD has excelled in producing highly specific agents for validated targets, though it remains constrained by existing biological knowledge and traditionally "druggable" target classes.
The Hybrid Framework: Integrating PDD and TDD
Conceptual Foundation and Scientific Rationale
The hybrid PDD-TDD framework represents a strategic integration where phenotypic screening identifies novel compounds with therapeutic potential, followed by target-based approaches to elucidate mechanisms and optimize candidates. This synergistic model creates a virtuous cycle where each approach addresses the limitations of the other. PDD enables exploration of novel biology without target preconceptions, while TDD provides the mechanistic understanding necessary for rational optimization and safety profiling [54].
This hybrid approach is particularly valuable for addressing the challenge of "undruggable" targets—proteins traditionally considered inaccessible to small molecule therapeutics. The human proteome comprises approximately 20,000 proteins, with about 12,000 identified as playing roles in human diseases. Despite this, only approximately 10% of potential drug targets have been targeted by FDA-approved drugs, leaving a substantial majority without therapeutic interventions [54]. Hybrid approaches can overcome traditional limitations such as lack of binding sites, protein-protein interactions, or transient binding pockets that have rendered many targets "undruggable" through TDD alone [54].
BridGene's IMTAC Platform: A Case Study in Hybrid Integration
BridGene's chemoproteomic platform IMTAC exemplifies the hybrid PDD-TDD approach, systematically merging the benefits of both strategies [54]. The platform consists of three integrated components:
- Designing and synthesizing a high-quality library of covalent small molecules
- Screening against the entire proteome of live cells, where small molecules selectively bind to structurally matching protein pockets and form covalent bonds
- Qualitative and quantitative mass spectrometry analysis to identify interacting proteins and quantify binding strengths [54]
This platform has demonstrated remarkable success, identifying small molecule ligands for over 4,000 proteins, with approximately 75% of these targets lacking known ligands prior to discovery [54]. The coverage includes traditionally "undruggable" proteins such as transcription factors, epigenetic regulators, splicing factors, and E3 ligases.
Table 2: Key Research Reagent Solutions for Hybrid PDD-TDD Approaches
| Reagent/Technology | Function in Hybrid Approach | Application Examples |
|---|---|---|
| Covalent Small Molecule Libraries | Form enduring covalent bonds with challenging protein pockets; enable targeting of shallow or transient binding sites [54] | Targeting KRAS G12C; historically "undruggable" targets [54] |
| CETSA (Cellular Thermal Shift Assay) | Validate direct target engagement in intact cells and tissues; bridge biochemical potency and cellular efficacy [41] | Quantifying drug-target engagement of DPP9 in rat tissue [41] |
| IMTAC Platform | Simultaneously explore entire proteome; discover small molecule ligands for diverse targets including "undruggable" proteins [54] | Identified ligands for 4,000+ proteins; 75% without prior known ligands [54] |
| PROTACs | Enable targeted protein degradation by bringing target protein together with E3 ligase; exploit covalent libraries for covalent PROTACs [54] | Over 80 PROTAC drugs in development pipeline; expanding beyond cereblon, VHL to new ligases [37] |
| AI/ML Platforms | Screen compound libraries, predict protein structures, identify host-virus interaction networks; accelerate hit identification [37] | Machine learning boosting hit enrichment by 50-fold; AI-designed antibiotics against resistant strains [37] [41] |
The following diagram illustrates the integrated workflow of a hybrid PDD-TDD approach using the IMTAC platform as an exemplar:
Integrated PDD-TDD Workflow via Chemoproteomics
Experimental Protocols and Methodologies
Covalent Library Design and Live-Cell Screening Protocol
The foundation of successful hybrid discovery lies in the design and implementation of comprehensive screening strategies. The following protocol outlines key steps for covalent library design and live-cell screening:
Library Design Criteria:
- Incorporate diverse covalent warheads (e.g., acrylamides, α,β-unsaturated carbonyls) targeting nucleophilic amino acids
- Balance lipophilicity and molecular weight to maintain cellular permeability
- Include structural motifs known to engage protein families of interest (e.g., kinase hinge binders)
- Ensure chemical stability under physiological conditions [54]
Live-Cell Screening Procedure:
- Culture relevant cell lines under physiological conditions (37°C, 5% CO₂)
- Treat with covalent library compounds across concentration range (typically 1 nM - 10 μM)
- Incubate for appropriate duration (4-24 hours) to allow binding equilibrium
- Wash cells to remove unbound compounds
- Lyse cells and digest proteins with trypsin
- Process peptides for mass spectrometry analysis [54]
Critical Considerations:
- Maintain cell viability throughout experiment (>90% by ATP assay)
- Include controls for non-specific binding (DMSO vehicle)
- Use isobaric mass tags (TMT) for multiplexed quantitative comparisons
- Employ affinity enrichment where appropriate for low-abundance targets [54]
Target Deconvolution and Validation Workflow
Following phenotypic screening and proteome-wide binding assessment, systematic target deconvolution and validation are essential:
Mass Spectrometry Analysis:
- Perform LC-MS/MS on tryptic peptides using high-resolution instrument (Orbitrap Fusion Lumos or similar)
- Use data-dependent acquisition for protein identification
- Employ isobaric labeling (TMT) for quantitative comparison between treatment groups
- Analyze data using search engines (MaxQuant, Spectronaut) against appropriate proteome database
- Apply significance thresholds (fold-change >2, p-value <0.05) for hit calling [54]
Orthogonal Validation:
- CETSA: Measure thermal stability shifts of putative targets in intact cells
- Treat cells with compound or vehicle control
- Heat cells across temperature gradient (37-65°C)
- Lysate cells and quantify soluble protein by immunoblot or MS
- Calculate ΔTm (shift in melting temperature) [41]
- Cellular Target Engagement:
- Use cellular assays measuring downstream pathway modulation
- Employ CRISPRi/CRISPRa to validate target necessity
- Implement resistance mutation studies to confirm on-target activity
- Functional Validation:
- Assess phenotype reversal with target-specific tools (siRNA, CRISPR)
- Establish correlation between target occupancy and phenotypic response [8]
Emerging Technologies Enhancing Hybrid Approaches
AI-Powered Integration and Analysis
Artificial intelligence has emerged as a powerful enabler of hybrid PDD-TDD strategies, with machine learning models now routinely informing target prediction, compound prioritization, and pharmacokinetic property estimation [41]. Recent advances demonstrate that integrating pharmacophoric features with protein-ligand interaction data can boost hit enrichment rates by more than 50-fold compared to traditional methods [41]. AI-powered "virtual patient" platforms and quantitative systems pharmacology (QSP) models simulate thousands of individual disease trajectories, allowing teams to test dosing regimens and refine inclusion criteria before clinical stages [37].
Covalent Chemoproteomics for "Undruggable" Targets
Covalent drugs have emerged as a powerful strategy for addressing limitations of traditional non-covalent therapies, particularly for challenging targets [54]. Their mechanism offers several transformative advantages:
- Higher Biochemical Efficiency: Irreversibility translates to potent and sustained effects
- Stronger & More Persistent Effects: Stable covalent bonds enable prolonged target modulation
- Reduced Dosage & Frequency: Enduring impact permits lower, less-frequent dosing
- Separation of Pharmacokinetics and Pharmacodynamics: Clearer distinction between drug clearance and pharmacological action
- Potential to Prevent Drug Resistance: Irreversible modification can thwart resistance development [54]
Covalent drugs particularly excel at targeting protein-protein interactions, disease-driving mutant proteins, and redox-regulatory proteins—target classes traditionally considered "undruggable" [54].
The integration of phenotypic and target-based drug discovery represents a maturation of the drug development field, moving beyond ideological debates toward pragmatic solutions that leverage the strengths of both approaches. As hybrid methodologies continue to evolve, several key trends are likely to shape their future implementation:
First, the increasing sophistication of human-based phenotypic platforms—including microphysiological systems, organ-on-chip technologies, and complex coculture models—will enhance the physiological relevance of initial screening phases [8]. Second, advances in chemoproteomics, structural biology, and functional genomics will accelerate target deconvolution, historically a major bottleneck in PDD [54]. Third, AI and machine learning will increasingly integrate diverse data types (genomic, proteomic, phenotypic) to generate testable hypotheses about mechanism of action and optimize chemical matter [37] [41].
The collaboration between BridGene and Takeda Pharmaceuticals, focused on implementing a cutting-edge drug development approach that combines PDD and TDD for neurodegenerative diseases, exemplifies the growing recognition of hybrid strategies' potential [54]. As these approaches demonstrate success in addressing high-unmet medical needs, their adoption is likely to expand across the industry.
For researchers implementing hybrid strategies, success will depend on maintaining a balance between biological complexity and mechanistic understanding, leveraging the best of both phenotypic serendipity and target-focused precision. By embracing this integrated framework, the drug discovery community can systematically address the substantial portion of the human proteome that remains untargeted, bringing new hope to patients with diseases currently considered untreatable.
Critical Analysis and Future Directions: Weighing the Evidence for PDD and TDD
The pursuit of first-in-class (FIC) drugs, characterized by novel mechanisms of action, represents the vanguard of pharmaceutical innovation, aiming to deliver transformative treatments for challenging diseases. These drugs are considered the main drivers of new drug discovery, holding the key to addressing unmet medical needs through novel targets and mechanisms [55]. The central thesis of this analysis is that the origins of FIC drugs are profoundly shaped by the fundamental strategy employed in the discovery process: Phenotypic Drug Discovery (PDD) versus Target-Based Drug Discovery (TDD). A meta-analysis of clinical studies in acute myeloid leukemia (AML) provides evidence-based support for PDD, indicating it offers benefits over TDD, including lower clinical failure rates [56]. This whitepaper provides a comprehensive quantitative and qualitative analysis of FIC drug success rates, framed within the PDD vs. TDD paradigm, to guide researchers, scientists, and drug development professionals in optimizing their discovery strategies.
Quantitative Landscape of Drug Development Success
Understanding the broader context of clinical development success rates (ClinSR) is crucial for evaluating the performance of FIC drugs. A large-scale dynamic analysis of clinical development programs from 2001 to 2023, involving 20,398 programs and 9,682 molecular entities, reveals critical trends and benchmarks [57] [58].
Table 1: Dynamic Clinical Trial Success Rates (ClinSR) in the 21st Century [57] [58]
| Category | Overall Success Rate | Key Trends and Variations |
|---|---|---|
| Global ClinSR (2001-2023) | Varies 7%-20% (varies by study) | Declined since early 21st century, hit a plateau, and recently started to increase. |
| Drug Repurposing | Lower than that for all drugs in recent years | Active strategy in the past two decades, but with an unexpectedly low recent ClinSR. |
| Anti-COVID-19 Drugs | Extremely low ClinSR | Highlights challenges in rapid development for novel pathogens. |
| Disease Areas | Great variation | Success rates differ significantly across various therapeutic areas. |
| Drug Modalities | Great variation | Success rates differ significantly across different drug types. |
Table 2: Recent First-in-Class Drug Approvals and Characteristics (2023-2024) [55]
| Characteristic | Distribution (2023-2024) | Noteworthy Examples |
|---|---|---|
| Molecule Type | Small-Molecule Drugs: 51.9%Macromolecule Drugs: 48.1% | Small molecules illustrate new chemical entities; macromolecules (mainly antibodies) show a growing trend. |
| Top Indication | Cancer (22.0%) | 18 FIC therapies approved, revealing high patient need and intense R&D focus. |
| Target Innovation | Diverse enzymes most common (32.1%) | 26 novel targets identified, with kinases providing more pioneering targets for FIC drugs. |
The data indicates that oncology remains the top priority for FIC drug development, accounting for 22% of approvals in 2023-2024 [55]. This is consistent with broader FDA approval trends, which show a significant surge in accelerated approvals for oncology drugs, particularly targeted therapies and immunotherapies [59]. Furthermore, the high proportion of macromolecule drugs, predominantly antibodies, among recent FIC approvals underscores the impact of new biotechnology techniques on drug discovery [55].
Phenotypic vs. Target-Based Drug Discovery: A Comparative Analysis
The choice between PDD and TDD is a fundamental strategic decision in the early stages of drug discovery. Evidence from a meta-analysis of 2918 clinical studies involving 466 unique drugs for Acute Myeloid Leukemia (AML) provides quantitative support for the advantages of the phenotypic approach [56].
Table 3: PDD vs. TDD: Evidence from an AML Meta-Analysis [56]
| Parameter | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| Clinical Failure Rate | Lower | Higher |
| Primary Reason for Failure | Fails less often due to a lack of efficacy. | - |
| Adaptation of Drugs | Provides an advantage in adapting drugs from other clinical indications. | - |
| Recommended Use | Recommended for future oncology drug discovery based on evidence. | - |
The core distinction lies in the starting point of the discovery campaign. Target-Based Drug Discovery (TDD) begins with a hypothesis about a specific molecular target's role in a disease. The subsequent screening process is designed to identify compounds that modulate this predefined target. In contrast, Phenotypic Drug Discovery (PDD) starts with observing a desired therapeutic effect in a complex cellular or whole-organism system, without a preconceived notion of the specific molecular target. The molecular mechanism of action is elucidated later.
The following workflow delineates the strategic decision-making process and the divergent paths of PDD and TDD.
Experimental Protocols for FIC Drug Discovery
This section outlines detailed methodologies for key experiments in both PDD and TDD frameworks, crucial for identifying high-quality hits and validating their potential as FIC candidates.
Protocol 1: High-Confidence Hit Identification in Phenotypic Screening
This protocol is designed to move beyond simple activity readouts and identify compounds with a higher probability of translational success by incorporating early target engagement assessment [56] [60].
- Objective: To identify compounds that induce a relevant phenotypic response in a physiologically relevant model and directly engage the intended molecular target within the cell.
- Materials:
- Physiologically Relevant Cell Model: Primary cells or engineered cell lines that accurately reflect the disease biology.
- Compound Library: A diverse collection of small molecules or biologics.
- Phenotypic Assay Reagents: Antibodies for immunostaining, fluorescent dyes, or other probes to quantify the phenotypic endpoint (e.g., cell differentiation, protein aggregation).
- CETSA Reagents [60]: Lysis buffer, protease inhibitors, and equipment for Western Blot, ELISA, or mass spectrometry to measure target protein stability.
- Procedure:
- Phenotypic Screening: Plate cells in 384-well plates. Treat with compounds from the library at a single concentration (e.g., 10 µM) for a predetermined period (e.g., 48 hours). Induce the disease-relevant phenotype if necessary.
- Phenotypic Readout: Fix and stain cells to measure the phenotypic endpoint using high-content imaging or a plate reader. Normalize data to positive and negative controls.
- Hit Triage: Select compounds that show significant activity in the phenotypic assay (>3 standard deviations from negative control).
- Target Engagement Validation (CETSA): [60]
- Treat separate aliquots of the same cell model with DMSO (vehicle) or selected hit compounds.
- Heat the cell aliquots to a range of different temperatures (e.g., 50°C to 65°C).
- Lyse the cells and isolate the soluble protein fraction.
- Quantify the amount of intact, soluble target protein remaining at each temperature using an immunoassay or mass spectrometry.
- Data Analysis: Compounds that cause a shift in the thermal stability of the target protein (indicating direct binding) are classified as high-confidence hits. Prioritize these for lead optimization.
Protocol 2: AI-Driven Virtual Screening for Target-Based Discovery
This protocol leverages artificial intelligence to efficiently screen ultra-large virtual compound libraries against a defined protein target, accelerating the identification of novel chemical starting points [61].
- Objective: To computationally screen millions of compounds to identify a focused set of high-probability binders for a validated disease target.
- Materials:
- Target Structure: A high-resolution 3D structure of the target protein from X-ray crystallography, Cryo-EM, or a high-quality predicted model from AlphaFold2.
- Virtual Compound Libraries: Databases of commercially available or make-on-demand compounds (e.g., ZINC20, Enamine REAL).
- Computational Infrastructure: High-performance computing (HPC) cluster or cloud computing platform with GPU acceleration.
- AI/ML Software: Molecular docking software (e.g., AutoDock Vina, Glide), and/or a pre-trained generative AI model for de novo molecular design.
- Procedure:
- Target Preparation: Prepare the protein structure by adding hydrogen atoms, assigning protonation states, and defining the binding site grid.
- Library Preparation: Download and curate the virtual compound libraries, generating relevant tautomers and protonation states at physiological pH.
- AI-Powered Screening:
- Option A (Structure-Based Virtual Screening): Perform molecular docking of the entire library against the target's binding site. Use a scoring function to rank compounds by predicted binding affinity.
- Option B (Generative AI Design): Use a generative AI model trained on known binders and chemical libraries to propose novel molecular structures optimized for the target's binding pocket.
- Hit Selection and Filtering: Apply drug-like filters (e.g., Lipinski's Rule of Five, PAINS filters) to the top-ranking compounds. Cluster compounds by structural similarity to select a diverse set of 100-500 compounds for experimental testing.
- Experimental Validation: Procure the selected compounds and test them in a biochemical binding or functional assay to confirm activity, thus validating the AI predictions.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagent Solutions for FIC Drug Discovery Experiments
| Research Reagent / Material | Function in Discovery Workflow |
|---|---|
| Physiologically Relevant Cell Models | Provides a disease-relevant cellular context for phenotypic screening and early efficacy/toxicity assessment. |
| CETSA Reagents [60] | Enables direct, label-free measurement of intracellular target engagement in live cells, validating mechanistic hypotheses. |
| AI/ML Software & Models [61] | Accelerates target identification, virtual screening, and de novo molecular design by analyzing complex biological and chemical datasets. |
| High-Content Imaging Systems | Automates the quantitative analysis of complex phenotypic changes in cells (morphology, protein localization, etc.). |
| Gene Editing Tools (e.g., CRISPR-Cas9) | Enables target validation and the creation of isogenic cell lines for mechanistic studies. |
The Impact of AI and Emerging Technologies
Artificial Intelligence is fundamentally transforming the FIC drug discovery process. AI-discovered drugs are reported to show significantly higher success rates in Phase I trials (80-90%) compared to traditionally developed drugs (40-65%) [61]. This improvement stems from AI's ability to enhance predictive accuracy and reduce costly late-stage failures.
AI's impact is felt across the pipeline:
- Target Identification: AI analyzes genomic and proteomic data to uncover novel disease targets and predict their druggability [61].
- Virtual Screening: Machine learning models can screen millions of compounds in silico in a fraction of the time of traditional HTS, identifying promising candidates with optimized properties [61].
- Lead Optimization: AI predicts ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties early, guiding the selection of safer, more effective leads [61].
- Drug Repurposing: AI analyzes vast datasets to identify new therapeutic uses for existing drugs, offering a faster, lower-risk path to new indications [62] [61].
The following diagram illustrates how AI integrates into and enhances the core PDD and TDD workflows.
The quantitative and qualitative analysis presented herein demonstrates that the origin of first-in-class drugs is intrinsically linked to the discovery strategy. The evidence strongly supports Phenotypic Drug Discovery (PDD) as a robust strategy for generating FIC drugs with a lower likelihood of clinical failure due to efficacy [56]. This advantage likely stems from PDD's focus on observable therapeutic effects in biologically complex systems from the outset. Conversely, Target-Based Drug Discovery (TDD) provides a focused, mechanism-driven approach that continues to yield successes, particularly as technologies for target validation improve.
The future of FIC drug discovery lies not in choosing one strategy exclusively, but in the strategic integration of both PDD and TDD principles, powerfully augmented by emerging technologies. The integration of AI and machine learning across the discovery pipeline is already dramatically accelerating timelines and improving success rates [61]. Furthermore, rigorous early-stage validation techniques, such as cellular target engagement assays [60], are de-risking the transition from hit to clinic. By leveraging these tools and understanding the quantitative success landscapes, drug development professionals can systematically navigate the complexities of FIC drug discovery to deliver the innovative therapies of tomorrow.
The process of drug discovery has long been dominated by two distinct philosophies: Phenotypic Drug Discovery (PDD) and Target-Based Drug Discovery (TDD). TDD, a reductionist approach, begins with a hypothesized molecular target with a known or presumed function in the disease process, followed by screening for compounds that modulate this specific target [2]. In contrast, PDD is an empirical, biology-first strategy that relies on chemical interrogation of a disease-relevant biological system in a molecular-target-agnostic fashion [2]. Rather than focusing on a predefined target, PDD seeks compounds that produce a desirable therapeutic effect in a realistic disease model, with the molecular mechanism of action being elucidated later [8].
The significance of PDD has been highlighted by the surprising observation that a majority of first-in-class drugs approved between 1999 and 2008 were discovered empirically without a pre-specified target hypothesis [2]. This historical success, combined with modern technological advances, has fueled a major resurgence of PDD over the past decade. Modern PDD serves as a powerful engine for expanding the "druggable genome" – the subset of genes and biological pathways that can be targeted by therapeutic compounds. By focusing on disease phenotypes rather than preconceived molecular targets, PDD reveals novel targets and unexpected mechanisms of action (MoA) that would likely remain undiscovered through purely target-based approaches [2].
The PDD Advantage: Unlocking Novel Biology and First-in-Class Medicines
Success Stories: From Phenotypic Screens to Approved Drugs
The power of PDD is best illustrated by its success in delivering transformative medicines for challenging diseases. The following case studies demonstrate how phenotypic approaches have identified drugs with unprecedented mechanisms.
Table 1: Notable Drug Discoveries Originating from Phenotypic Screens
| Drug | Disease | Key Target/Mechanism Identified Post-Discovery | Significance |
|---|---|---|---|
| Ivacaftor, Elexacaftor, Tezacaftor [2] | Cystic Fibrosis (CF) | CFTR channel gating (potentiators) and folding/trafficking (correctors) | First therapies to address the underlying cause of CF; combination therapy addresses 90% of patients [2]. |
| Risdiplam, Branaplam [2] | Spinal Muscular Atrophy (SMA) | SMN2 pre-mRNA splicing modifier | First oral disease-modifying therapy for SMA; stabilizes the U1 snRNP complex, an unprecedented target [2]. |
| Daclatasvir [2] | Hepatitis C Virus (HCV) | HCV NS5A protein inhibitor | Discovered via an HCV replicon phenotypic screen; NS5A has no known enzymatic activity [2]. |
| Lenalidomide [2] | Multiple Myeloma | Binds Cereblon E3 ligase, degrading transcription factors IKZF1/IKZF3 | MoA elucidated years post-approval; pioneered the field of targeted protein degradation [2]. |
| SEP-363856 [2] | Schizophrenia | Novel MoA (unrelated to D2 antagonism) | Clinical-stage compound demonstrating PDD's ability to find new biology for complex CNS disorders [2]. |
Quantitative Advantages of the Phenotypic Approach
The impact of PDD is not merely anecdotal. Analysis of drug discovery outcomes reveals a clear, quantitative advantage for identifying first-in-class therapies. A seminal study covering 1999-2008 found that PDD was the source of a majority of these pioneering drugs [2]. This disproportionate success rate is the primary driver for the renewed interest in phenotypic strategies.
PDD excels in several key areas that contribute to this success:
- Expansion of Druggable Target Space: PDD has revealed that "druggability" extends beyond traditional target classes like enzymes and receptors. It has successfully targeted unexpected cellular processes, including pre-mRNA splicing, protein folding, trafficking, and degradation [2].
- Revealing Novel Mechanisms of Action (MoA): As seen with lenalidomide and risdiplam, PDD can uncover entirely new biological principles for pharmaceutical intervention, creating new fields of drug discovery in the process [2].
- Embracing Polypharmacology: Many effective drugs act on multiple targets. PDD is inherently suited to identify compounds where the therapeutic effect arises from a synergistic combination of targets (on-target polypharmacology), which is particularly valuable for complex, polygenic diseases [2].
Core Methodologies in Modern Phenotypic Drug Discovery
The modern PDD workflow integrates advanced disease models with sophisticated target deconvolution techniques to move systematically from a phenotypic hit to a validated drug candidate.
The PDD Workflow: From Screening to Target Identification
The following diagram outlines the core stages of a phenotypic drug discovery campaign, highlighting its iterative and biology-centric nature.
Experimental Protocols for Target Deconvolution
A critical challenge in PDD is the identification of a compound's molecular target(s)—a process known as target deconvolution. "Unmodified methods," which do not require chemical alteration of the drug, have become essential tools as they reduce non-specific interactions and false positives by using the drug in its native state [63]. The table below details key unmodified methodologies.
Table 2: Unmodified Methodologies for Target Identification in PDD
| Method | Core Principle | Key Steps (Protocol Summary) | Advantages | Limitations |
|---|---|---|---|---|
| DARTS [63] | Drug binding protects the target protein from proteolysis. | 1. Incubate cell lysate with/without drug.2. Perform limited proteolysis (e.g., with thermolysin).3. Analyze by SDS-PAGE or MS; bands/proteins diminished in drug sample are potential targets. | Simple, low-cost; no special equipment beyond a mass spectrometer. | Can miss targets that do not show proteolysis resistance. |
| CETSA [63] | Drug binding stabilizes the target protein against heat-induced denaturation. | 1. Treat intact cells or lysate with/without drug.2. Heat to different temperatures.3. Centrifuge to separate soluble (native) from insoluble (denatured) protein.4. Quantify soluble target protein (e.g., via Western blot or MS). | Can be performed in intact cells, revealing cellular engagement. | Requires a specific antibody or MS method for detection. |
| LiP-MS [63] | Drug binding alters protein structure, changing its protease digestion pattern. | 1. Treat cell lysate with/without drug.2. Digest with a non-specific protease (e.g., proteinase K).3. Analyze peptides by mass spectrometry.4. Identify proteins with shifted proteolytic patterns. | Can screen the entire proteome without preprocessing. | Complex data analysis; requires specialized bioinformatics. |
| SPROX [63] | Drug binding increases protein stability against chemical denaturation. | 1. Treat cell lysate with/without drug.2. Expose to a series of denaturant concentrations (e.g., guanidine-HCl).3. Measure the rate of methionine oxidation by MS.4. Identify proteins with shifted denaturation curves. | Can be performed on complex mixtures. | Limited to proteins containing methionine. |
| MSIPP [63] | Drug binding increases protein stability against mechanical stress-induced denaturation. | 1. Incubate protein extract with/without drug.2. Apply mechanical stress (e.g., by eddying with particles).3. Centrifuge and analyze the supernatant; increased protein in the drug sample indicates binding. | Novel mechanism; does not rely on heat or chemicals. | Less established; requires further validation. |
The following diagram illustrates the shared thermodynamic principle underlying many of these stability-based methods and their experimental workflows.
The Scientist's Toolkit: Essential Research Reagents
Implementing the aforementioned protocols requires a suite of specialized research reagents and tools. The following table catalogues the essential components for a typical PDD target identification pipeline.
Table 3: Key Research Reagent Solutions for PDD Target Identification
| Reagent / Tool | Function in PDD | Specific Application Examples |
|---|---|---|
| Disease-Relevant Cell Models | Provide the biological context for the initial phenotypic screen and subsequent MoA studies. | Primary human cells, induced pluripotent stem cell (iPSC)-derived models, microphysiological systems ("organ-on-a-chip") [8]. |
| Phenotypic Assay Kits | Enable high-throughput measurement of disease-relevant phenotypes. | Apoptosis, cell proliferation, neurite outgrowth, cytokine release, and viral replication assay kits. |
| Non-Specific Proteases | Used in DARTS and LiP-MS to probe for drug-induced protein stability and conformational changes. | Thermolysin, Proteinase K, subtilisin [63]. |
| Chemical Denaturants | Used in SPROX to measure protein folding stability in the presence and absence of a drug. | Guanidine hydrochloride (GdnHCl), urea [63]. |
| Mass Spectrometry Systems | The core analytical platform for proteome-wide identification and quantification of target proteins. | High-resolution LC-MS/MS systems for LiP-MS, SPROX, and CETSA workflows [63]. |
| CRISPR Screening Libraries | Enable genome-wide functional screening to identify genes involved in a drug's mechanism of action or resistance. | Genome-wide sgRNA libraries for identifying genetic modifiers of drug sensitivity [40]. |
Integrating PDD with Cutting-Edge Technologies
The future of PDD lies in its integration with other powerful technological platforms, which help address its traditional challenges, such as target deconvolution and the use of complex disease models.
Functional Genomics and CRISPR Screening: CRISPR-Cas9 screening technology provides a precise and scalable platform for functional genomics. The development of extensive single-guide RNA (sgRNA) libraries enables high-throughput screening that systematically investigates gene-drug interactions across the entire genome [40]. This approach is particularly powerful in PDD for identifying genes whose loss-of-function either reverses or enhances the phenotypic effect of a drug, thereby revealing its mechanism of action or potential resistance pathways. CRISPR screening has been broadly applied in cancer, infectious diseases, and neurodegenerative conditions to elucidate drug mechanisms [40].
Advanced Disease Models: There is a growing shift toward more physiologically relevant models in PDD. This includes the use of human primary cells, co-culture systems, and microphysiological systems (organoids and "organs-on-a-chip") that better recapitulate the tissue and disease microenvironment [8]. When combined with CRISPR, organoid-based screening enables highly efficient and physiologically relevant drug target identification [40].
Artificial Intelligence and Big Data: The large, complex datasets generated by phenotypic screens, proteomics (e.g., from LiP-MS/CETSA), and functional genomics require sophisticated computational tools. Machine learning and AI are increasingly used to analyze these datasets, identify patterns, and predict both drug targets and potential efficacy or toxicity [40]. Integrating PDD data with other 'omics' data sources fuels a deeper understanding of drug efficacy and toxicity mechanisms, supporting clinical strategies through pathway-based decision frameworks [8].
Phenotypic Drug Discovery has firmly re-established itself as a powerful, productive, and necessary approach in the modern drug discovery landscape. By focusing on therapeutic effects in biologically complex and disease-relevant systems, PDD bypasses the constraints of pre-defined target hypotheses, enabling the serendipitous discovery of novel biology and first-in-class medicines. Its unique ability to expand the druggable genome—revealing new target classes and unexpected mechanisms of action—makes it an indispensable complement to target-based approaches.
The continued success and evolution of PDD are being driven by advancements in key areas: the development of more human-relevant disease models, the maturation of powerful "unmodified" target deconvolution methods like CETSA and LiP-MS, and integration with transformative technologies such as functional genomics (CRISPR) and artificial intelligence. As these tools and methods continue to mature, PDD is poised to systematically uncover the next generation of therapeutic targets and deliver innovative treatments for diseases with high unmet medical need.
For much of the past century, drug discovery was dominated by the "one target–one drug" paradigm, which focused on developing highly selective ligands as "magic bullets" for individual disease proteins [64]. This strategy was predicated on the belief that maximal specificity would yield optimal therapeutic benefit while minimizing off-target effects. However, the reductionist oversight of the complex, redundant, and networked nature of human biology has led to significant limitations, with approximately 90% of single-target candidates failing in late-stage trials due to lack of efficacy or unexpected toxicity [64]. In contrast, phenotypic drug discovery (PDD) approaches, which rely on therapeutic effects observed in realistic disease models without a predefined molecular target hypothesis, have proven to be a more successful strategy for discovering first-in-class medicines [2] [49]. This article reexamines polypharmacology—the rational design of compounds to interact with multiple targets—within the broader context of PDD versus target-based drug discovery (TDD), highlighting how multi-target strategies address the limitations of excessive specificity in treating complex diseases.
Theoretical Foundations: From Magic Bullets to Network Therapeutics
The Biological Rationale for Polypharmacology
Biological systems exhibit remarkable resilience to single-point perturbations through compensatory mechanisms and redundant functions [65]. Disease often emerges from the breakdown of robust physiological systems due to multiple genetic and/or environmental factors, leading to the establishment of robust disease conditions that are frequently refractory to single-target interventions [65]. The network nature of human biology means that modulating a lone node in a complex network can easily be circumvented by the system, resulting in lack of long-term efficacy or emergence of resistance [64].
Table 1: Comparative Analysis of Drug Discovery Paradigms
| Parameter | Target-Based Discovery (TDD) | Phenotypic Discovery (PDD) | Polypharmacology |
|---|---|---|---|
| Starting Point | Hypothesis about specific target | Disease phenotype or biomarker | Multiple targets or phenotypic screen |
| Success Rate (First-in-Class) | Lower | Higher (~61% of first-in-class drugs, 1999-2008) [2] | Emerging evidence of success |
| Target Space | Limited to known, druggable targets | Unrestricted, can reveal novel biology | Designed for multiple predefined or discovered targets |
| Chemical Design | Optimized for single-target selectivity | Unbiased by target constraints | Balanced multi-target engagement |
| Therapeutic Application | Single-gene disorders | Complex, multifactorial diseases | Complex diseases, drug resistance |
| Clinical Translation | Often poor correlation between in vitro and in vivo efficacy [65] | Better predictive value of complex models | Addresses network-level disease mechanisms |
Limitations of Excessive Specificity
The traditional preference for highly specific drugs was driven by safety concerns—the desire to minimize "off-target" interactions that could cause side effects. Paradoxically, many effective medications were later found to be "promiscuous" in their action, hitting multiple targets [64]. Excessive specificity presents several critical limitations:
- Therapeutic Resistance: Pathogens and cancer cells frequently develop resistance to highly specific drugs through single-point mutations in the drug's target [64]. This is particularly problematic in antimicrobial chemotherapy and oncology.
- Insufficient Efficacy in Complex Diseases: In multifactorial conditions such as neurodegenerative disorders, metabolic diseases, and psychiatric illnesses, single-target interventions often fail to address the complex disease network [65] [64].
- Biological Redundancy and Compensation: Biological systems can activate alternative pathways when a single pathway is perturbed, leading to diminished therapeutic effect over time [64].
Polypharmacology in Clinical Applications
Disease Areas Benefiting from Multi-Target Approaches
Table 2: Multi-Target Drug Applications Across Disease Areas
| Disease Area | Challenge with Single-Target Therapy | Multi-Target Solution | Representative Agents |
|---|---|---|---|
| Neurodegenerative Disorders (Alzheimer's, Parkinson's) | Multiple pathological processes (protein aggregation, oxidative stress, neuroinflammation) | Multi-target-directed ligands (MTDLs) integrating cholinesterase inhibition, anti-amyloid, and antioxidant activities [64] | Memoquin (AChE inhibition, anti-amyloid, antioxidant) [64] |
| Oncology | Tumor heterogeneity, resistance mechanisms, redundant signaling pathways | Multi-kinase inhibitors, antibody-drug conjugates, bispecific antibodies [64] [66] | Imatinib (BCR-ABL, c-KIT, PDGFR) [2]; Sorafenib (multiple kinases) [64] |
| Metabolic Disorders (Type 2 diabetes, obesity) | Multiple interconnected abnormalities requiring polypharmacy | Dual and triple agonists engaging related metabolic pathways [64] [66] | Tirzepatide (GLP-1/GIP receptor agonist) [64] [66] |
| Infectious Diseases | Rapid development of antimicrobial resistance | Single molecules attacking multiple bacterial targets or host factors [64] | Antibiotic hybrids (e.g., quinolone + membrane disruptor) [64] |
| Psychiatric Disorders (depression, schizophrenia) | Complex etiology involving multiple neurotransmitter systems | Compounds with balanced activity across several receptor systems [65] | SEP-363856 (novel mechanism discovered via PDD) [2] |
Recent Clinical Successes (2023-2024)
The continued relevance of polypharmacology is evidenced by recent drug approvals. Analysis of drugs approved in 2023-2024 in Germany revealed that 18 of 73 newly introduced substances (approximately 25%) align with the polypharmacology concept [66]. These include:
- 10 antitumor agents including antibody-drug conjugates (e.g., loncastuximab tesirine targeting CD19 plus cytotoxic payload), bispecific antibodies, and multi-kinase inhibitors
- 5 drugs for autoimmune/inflammatory disorders
- 1 antidiabetic/anti-obesity drug (tirzepatide, a dual GLP-1/GIP receptor agonist)
- 1 agent for hand eczema
- 1 modified corticosteroid [66]
These recent approvals demonstrate the pharmaceutical industry's continued investment in multi-target approaches across therapeutic areas, particularly in oncology where network-based interventions are most advanced.
Methodological Approaches: Integrating PDD and Rational Design
Experimental Strategies for Multi-Target Drug Discovery
Diagram 1: Integrated Drug Discovery Workflow. This diagram illustrates the convergence of phenotypic screening and rational design approaches in modern polypharmacology.
Research Reagent Solutions for Polypharmacology Studies
Table 3: Essential Research Tools for Multi-Target Drug Discovery
| Research Tool Category | Specific Examples | Function in Polypharmacology Research |
|---|---|---|
| Phenotypic Screening Platforms | Human organoids [67], Microphysiological systems [8], Primary cell co-cultures | Provide physiologically relevant models for target-agnostic compound screening and validation |
| Target Deconvolution Technologies | Connectivity Map (gene expression profiles) [65], CRISPR functional screens [64], Proteomic profiling | Identify mechanism of action for phenotypic hits and map compound-target networks |
| Computational Design Tools | AI-based generative models [64] [66], Molecular docking simulations [66], Network pharmacology algorithms [64] | Predict multi-target interactions, design novel MTDLs, identify synergistic target combinations |
| Structural Biology Resources | Protein structure databases, Molecular modeling software, X-ray crystallography facilities | Enable structure-based design of merged pharmacophores and optimization of target engagement |
| In Vivo Disease Models | Genetically engineered animal models, Patient-derived xenografts, Disease-specific phenotype models | Validate efficacy of multi-target compounds in complex biological systems |
Molecular Design Strategies for Multi-Target Ligands
The structural design of multi-target-directed ligands (MTDLs) follows three primary strategies, each with distinct advantages and challenges:
Linked Pharmacophores: Two distinct pharmacophores connected via a spacer (linker), which may be enzyme-degradable in vivo. Example: loncastuximab tesirine, an antibody-drug conjugate where the antibody (targeting CD19) is linked to the cytotoxic agent via a spacer [66].
Fused Pharmacophores: Direct attachment of pharmacophores via covalent bonding without linker groups. Example: tirzepatide, where specific amino acid residues for GLP-1 and GIP receptor engagement are fused [66].
Merged Pharmacophores: Integration of multiple pharmacophores into a single, unified structural entity. Example: sparsentan, where elements for ETA and AT1 receptor blockade overlap completely within the molecular architecture [66].
Each approach presents distinct challenges in medicinal chemistry. Linked pharmacophores often produce larger molecules with potential pharmacokinetic complications, while merged pharmacophores require sophisticated molecular design to maintain balanced activity at multiple targets without excessive molecular size [65].
Technological Advances Driving Polypharmacology
Artificial Intelligence and Computational Methods
Recent advances in artificial intelligence have dramatically accelerated multi-target drug discovery:
- Generative Models: AI-driven generative chemistry enables de novo creation of novel multi-target structures using deep learning frameworks [64] [66].
- Target Prediction: Machine learning algorithms screen compound libraries and predict multi-target interactions before synthesis [64].
- Network Pharmacology: Computational analysis of disease networks identifies synergistic target combinations and avoids antagonistic target pairs [64].
These approaches are particularly valuable for addressing the "key number 3" problem metaphorically described in traditional lock-and-key paradigm—the challenge of designing a single key (ligand) that can open multiple locks (targets) despite their structural differences [65].
Experimental Models for Validation
Modern PDD leverages increasingly sophisticated models that bridge the gap between traditional in vitro assays and clinical translation:
- Human Organoids: 3D organotypic cultures that recapitulate tissue-level complexity and disease phenotypes for more predictive screening [67].
- Microphysiological Systems: Organ-on-a-chip platforms that model human physiology and disease states for compound evaluation [8].
- Gene Expression Profiling: Tools like the Connectivity Map, which connects disease signatures and small molecules through gene expression profiles, enabling identification of multi-target mechanisms [65].
Diagram 2: Technology Convergence in MTDL Development. This diagram shows how artificial intelligence, advanced experimental models, and synthetic chemistry converge to enable rational design of multi-target therapeutics.
Challenges and Future Perspectives
Current Limitations in Polypharmacology
Despite promising advances, rational polypharmacology faces several significant challenges:
- Molecular Design Complexity: Designing molecules with balanced activity at multiple targets while avoiding undesirable off-target effects remains challenging [65] [66].
- Preclinical Validation Hurdles: The complexity of validating multi-target mechanisms in biologically relevant systems can be resource-intensive [68].
- Safety Concerns: Promiscuous compounds inherently carry higher risks for unexpected adverse events, as exemplified by historical cases like thalidomide [66].
- Regulatory Considerations: The pathway for approval of drugs with complex mechanisms of action is less defined than for single-target agents [66].
Emerging Opportunities and Future Directions
The field of polypharmacology continues to evolve with several promising trends:
- Expansion of Targeted Protein Degradation: PROTACs (PROteolysis TArgeting Chimeras) and molecular glues represent a novel form of polypharmacology that co-opts cellular machinery for protein degradation [37]. As of 2025, more than 80 PROTAC drugs are in development pipelines [37].
- Integration with Personalized Medicine: Multi-target approaches can be tailored to individual patient disease networks and genetic profiles [66].
- Advanced Biomarker Development: Blood-based and imaging biomarkers enable better patient stratification and assessment of multi-target drug efficacy, particularly in complex diseases like neurodegeneration [37].
- AI-Enhanced Clinical Trials: Quantitative systems pharmacology models and "virtual patient" platforms simulate disease trajectories and treatment responses, optimizing trial design for multi-target agents [37].
The reexamination of polypharmacology reveals a paradigm shift away from excessive specificity toward network therapeutics that embrace biological complexity. Multi-target drugs have demonstrated particular success in complex diseases where single-target approaches have repeatedly failed, with phenotypic drug discovery serving as a fertile source of novel multi-target mechanisms. The integration of artificial intelligence, advanced disease models, and structural biology has transformed polypharmacology from serendipitous discovery to rational design. As drug discovery confronts increasingly complex therapeutic challenges, the strategic integration of multi-target approaches within both phenotypic and target-based frameworks will be essential for delivering transformative medicines that address the network-based nature of human disease.
The long-standing dichotomy in pharmaceutical research, pitting target-based drug discovery (TBDD) against phenotypic drug discovery (PDD), is being fundamentally transformed by artificial intelligence (AI). For decades, TBDD—the hypothesis-driven approach of modulating a specific, known molecular target—dominated industrial drug discovery due to its straightforward mechanism and high throughput [69] [10]. Conversely, PDD—an empirical approach that identifies compounds based on their effects in disease-relevant biological systems without presupposing a target—has experienced a major resurgence. This resurgence was fueled by analyses showing that between 1999 and 2008, a majority of first-in-class medicines originated from phenotypic strategies [2] [10]. PDD excels at identifying novel mechanisms of action and probing the full complexity of disease biology, but it has historically faced challenges in target deconvolution and hit validation [69] [2].
AI platforms are now dissolving the traditional boundaries between these two paradigms. By integrating and interpreting massive, multimodal datasets, AI is addressing core weaknesses of each approach while amplifying their strengths. This technical analysis evaluates how leading AI platforms are deploying distinct technological strategies to impact both TBDD and PDD, creating a new, more integrated operating system for modern drug discovery [15] [34].
Core Analytical Framework: PDD vs. TBDD
Comparative Analysis of Discovery Paradigms
The table below summarizes the fundamental characteristics, strengths, and challenges of the PDD and TBDD paradigms, which form the basis for understanding AI's transformative role.
Table 1: Fundamental Characteristics of Phenotypic and Target-Based Drug Discovery
| Feature | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TBDD) |
|---|---|---|
| Core Principle | Identifies compounds based on effects in disease-relevant biological systems without a pre-specified target [2] [10] | Focuses on modulating a specific, known molecular target (e.g., enzyme, receptor) with a hypothesized link to disease [69] [10] |
| Primary Screening | Cell-based or whole-organism assays measuring complex phenotypes [2] | Target-specific biochemical or biophysical assays (e.g., binding, enzyme activity) [69] |
| Key Strength | Higher potential for first-in-class medicines with novel mechanisms; captures biological complexity and polypharmacology [69] [2] | Rational, straightforward mechanism; high-throughput capability; generally simpler optimization [69] |
| Key Challenge | Target deconvolution (identifying the mechanism of action) can be difficult and time-consuming [69] [2] | Relies on often imperfect assumptions about a target's link to disease, leading to translational failures [69] [10] |
| AI's Primary Value | Analyzing high-content data (e.g., imaging), deconvoluting mechanisms, and identifying novel biological insights [15] [34] | Rapidly designing and optimizing novel, drug-like molecules against a known target structure or data [15] |
The Resurgence of PDD and the Role of AI
The renewed interest in PDD is largely data-driven. Modern tools such as high-content imaging, single-cell technologies, and functional genomics (e.g., Perturb-seq) now allow for the capture of subtle, disease-relevant phenotypes at an unprecedented scale and resolution [2] [34]. This generates massive, information-rich datasets that are perfectly suited for AI and machine learning (ML) analysis. AI models can detect complex patterns in this data to identify active compounds, predict mechanisms of action, and even link phenotypic responses to underlying genomic, transcriptomic, or proteomic states [34]. This directly addresses the historical challenge of target deconvolution in PDD.
Leading AI Platforms and Their Strategic Approaches
The AI drug discovery landscape is populated by platforms with diverse technological specializations. The following table profiles leading companies, classifying their primary AI approach and impact on PDD and TBDD.
Table 2: Leading AI Drug Discovery Platforms and Their Impact on PDD and TBDD
| AI Platform / Company | Core AI Specialization & Technology | Impact on PDD | Impact on TBDD | Key Clinical/Preclinical Progress |
|---|---|---|---|---|
| Recursion | Phenomics-first systems: Maps high-content cellular images to genetic and chemical perturbations using ML [15]. | High; uses AI to extract disease-relevant features from complex phenotypic screens [15]. | Indirect; identifies novel targets via phenotypic screening for downstream TBDD. | Merged with Exscientia (2024) to integrate phenomics with generative chemistry [15]. |
| Exscientia | Generative chemistry & automated design: AI-driven small molecule design and optimization [15]. | Medium; incorporates patient-derived phenotypic data (e.g., via Allcyte acquisition) for compound validation [15]. | High; accelerates lead identification and optimization for known targets with fewer synthesized compounds [15]. | Multiple clinical candidates; e.g., LSD1 inhibitor (EXS-74539) in Phase I (2024) [15]. |
| Insilico Medicine | Integrated target-to-design pipeline: End-to-end AI from target discovery (PandaOmics) to molecule generation (Chemistry42) [15] [70]. | Medium; can initiate from genomic/transcriptomic data to propose novel targets based on disease biology. | High; fully AI-driven pipeline from novel target to generative molecule design. | ISM001-055 (TNK inhibitor for IPF) advanced from target to Phase I in 18 months; Phase IIa results in 2025 [15]. |
| Schrödinger | Physics-plus-ML design: Combines physics-based computational methods with machine learning [15]. | Lower; platform is predominantly structure-focused. | Very High; enables precise, structure-based drug design for computationally intensive targets. | Nimbus-originated TYK2 inhibitor, zasocitinib (TAK-279), advanced to Phase III trials [15]. |
| Atomwise | Deep learning for structure-based design: Uses convolutional neural networks (AtomNet) for molecular docking and virtual screening [70]. | Lower; primarily targets known protein structures. | Very High; enables virtual screening of trillion-compound libraries against protein targets. | Nominated an orally bioavailable TYK2 inhibitor as a development candidate (2023) [70]. |
| Ardigen | AI-powered phenotypic data analysis: PhenAID platform analyzes cell morphology data (e.g., Cell Painting) to identify MoA and bioactivity [34]. | Very High; specializes in interpreting high-content imaging to decode phenotypic complexity. | Medium; can predict on- and off-target activity of compounds from phenotypic data. | Used in collaborations to uncover new drug targets and refine lead compounds [34]. |
Analysis of Strategic Directions
The data reveals several key strategic directions in the AI-driven discovery landscape:
- Specialization vs. Integration: Some platforms, like Recursion and Ardigen, began with a deep focus on one paradigm (PDD) and are now integrating capabilities from the other. Others, like Insilico Medicine, built an integrated pipeline from the start. The merger of Recursion (phenomics) and Exscientia (generative chemistry) exemplifies a strategic move to create a full-stack, end-to-end AI discovery engine [15].
- Data as a Core Asset: The effectiveness of these platforms is directly tied to the quality and scale of their underlying data. Recursion's extensive phenomic database, BPGbio's access to a large multi-omics biobank, and BostonGene's integration of digital pathology with molecular data highlight that proprietary, multimodal data is a critical competitive advantage [70] [71].
- Validation through Clinical Progress: By 2025, over 75 AI-derived molecules had reached clinical stages, providing tangible proof of concept. While no AI-discovered drug has yet received full market approval, the progression of multiple candidates into Phase II and III trials indicates the field is moving beyond hype into a phase of tangible, clinical validation [15].
Technical Methodologies and Experimental Protocols
AI-Enhanced Phenotypic Screening Workflow
The following diagram illustrates a modern, AI-enhanced workflow for phenotypic drug discovery, integrating high-content screening with multi-omics data and AI analysis for target deconvolution.
This workflow leverages several key technologies and reagents to function effectively. The table below details the essential components of a modern, AI-driven phenotypic screening campaign.
Table 3: Key Research Reagents and Solutions for AI-Enhanced Phenotypic Screening
| Reagent / Solution | Function in Experimental Protocol |
|---|---|
| Patient-Derived Cells / 3D Organoids | Provides a biologically relevant, human-based model system that more accurately recapitulates disease biology compared to traditional cell lines [10]. |
| Cell Painting Assay Dyes | A panel of fluorescent dyes that stain multiple cellular components (nucleus, cytoplasm, mitochondria, etc.), generating a rich, high-content morphological profile for AI analysis [34]. |
| CRISPR Libraries | Enables genome-scale functional genomics screens (e.g., Perturb-seq) to link genetic perturbations to phenotypic outcomes, providing causal data for AI models [10] [34]. |
| Multi-Omics Reagents | Kits for RNA/DNA extraction, protein isolation, and metabolomic profiling are used to generate layered molecular data (transcriptomics, proteomics) that is integrated with phenotypic images [34] [71]. |
| Automated Liquid Handlers | Robotics (e.g., from Tecan, SPT Labtech) ensure consistent, high-throughput plating, dosing, and staining of cells, which is critical for generating reproducible, high-quality data for AI training [31]. |
AI-Driven Target-Based Design Workflow
For target-based discovery, AI platforms employ a different, highly automated workflow focused on rapid molecular design and optimization, as shown in the following diagram.
Case Studies of AI-Driven Discovery
Phenotypic Case Study: Recursion's Phenomics Platform
Recursion operates a highly automated, AI-driven phenomics platform. It conducts massive, parallelized phenotypic screens in human cell models, perturbing them with genetic tools (e.g., CRISPR) or small molecules. The platform uses high-content imaging to capture millions of cellular images daily, which are processed by ML models to extract quantitative feature vectors (a "phenomic profile") for each perturbation [15].
- AI Methodology: Deep learning models are trained to recognize subtle morphological patterns and associate them with known biological pathways, genetic perturbations, and disease states. This creates a massive map of biological relationships.
- Impact on PDD: This approach directly addresses the scale and deconvolution challenges of PDD. When a compound induces a phenotypic profile that clusters with profiles generated by known genetic perturbations (e.g., a kinase knockout), it provides a powerful, data-driven hypothesis for the compound's mechanism of action [15].
- Integration with TBDD: The platform outputs novel target hypotheses based on phenotypic associations, which can then be advanced into more traditional, AI-accelerated TBDD pipelines. The merger with Exscientia specifically aims to create a closed loop between phenotypic discovery and generative chemistry [15].
Target-Based Case Study: Insilico Medicine's End-to-End Pipeline
Insilico Medicine's platform provides a prime example of AI-driven TBDD. For their idiopathic pulmonary fibrosis (IPF) program, the process began with their target discovery engine, PandaOmics.
- AI Methodology for Target ID: PandaOmics uses AI to analyze massive volumes of multi-omics data, scientific literature, and clinical trial databases to identify and rank novel drug targets associated with a disease [15] [70].
- AI Methodology for Molecule Design: The top-ranked target (TNK) was then fed into their generative chemistry engine, Chemistry42. This system uses a combination of generative adversarial networks (GANs) and reinforcement learning to design novel molecular structures from scratch that are optimized for the target, along with key drug properties [15].
- Result: The entire process—from novel target identification to the generation of a candidate molecule (ISM001-055) ready for preclinical studies—was compressed to just 18 months, a fraction of the typical 3-5 year timeline. This candidate has since progressed into Phase II clinical trials [15].
Future Outlook and Synthesis
The convergence of PDD and TBDD, mediated by AI, points toward a future of integrative drug discovery. Key trends shaping this future include:
- The Rise of Foundation Models: Inspired by large language models, companies like Bioptimus are building universal AI foundation models for biology. Trained on massive genomic, proteomic, and cellular datasets, these models aim to capture the fundamental "rules" of biology, potentially enabling the prediction of therapeutic targets and drug responses with greater accuracy [72].
- Automation of the Discovery Lifecycle: The integration of AI design with automated robotic synthesis and testing, as seen with Exscientia's AutomationStudio and Iktos's robotics platform, is creating high-throughput, closed-loop systems. These systems can rapidly iterate through design-make-test-analyze cycles, accelerating optimization [15] [70].
- Data as the Ultimate Limiting Factor: Future progress will depend not only on better algorithms but also on access to high-quality, well-annotated, and multimodal datasets. Initiatives to standardize data (FAIR principles) and build large-scale, clinically annotated biobanks will be critical to training more powerful and predictive AI models [31] [34].
In conclusion, the AI revolution in drug discovery is not about one paradigm winning over the other. Instead, it is about creating a synergistic relationship between PDD and TBDD. AI platforms are the essential tools that translate the complex, unbiased findings from phenotypic screens into actionable, target-specific hypotheses, and then dramatically accelerate the process of designing and optimizing compounds against those targets. This synergy, powered by ever-improving AI, data, and automation, holds the promise of breaking Eroom's Law and delivering the next generation of life-changing medicines more efficiently.
The drug discovery process traditionally navigates between two principal strategies: Phenotypic Drug Discovery (PDD) and Target-Based Drug Discovery (TDD). Historically, drug discovery began with phenotypic observations, where compounds were selected based on their therapeutic effects on whole organisms or cells without knowledge of the specific biological target [2] [11]. With the advent of molecular biology and genomics in the 1980s, the industry pivoted to a more reductionist TDD approach, focusing on modulating specific, pre-validated molecular targets [2]. However, a landmark analysis revealing that a majority of first-in-class drugs approved between 1999 and 2008 were discovered through PDD has driven a major resurgence of this approach over the past decade [2] [9].
This whitepaper provides an in-depth, technical comparison of PDD and TDD, framing them not as opposing strategies but as complementary tools in the modern drug developer's arsenal. It is structured to guide researchers, scientists, and drug development professionals in selecting the optimal strategy for their specific project goals, leveraging recent successes, and integrating modern technological advancements like Artificial Intelligence (AI).
Core Concept and Workflow Comparison
Defining the Approaches
Phenotypic Drug Discovery (PDD) is defined as an approach that identifies bioactive compounds by their ability to modulate a disease-relevant phenotype in a cell, tissue, or whole organism system, without a pre-specified hypothesis about the molecular target [2] [11]. The focus is on the therapeutic effect in a realistic disease model, making it a biology-first, empirical strategy.
Target-Based Drug Discovery (TDD) is a hypothesis-driven approach that begins with the selection of a specific molecular target (e.g., a protein, enzyme, or receptor) believed to play a critical role in the disease pathogenesis. Compounds are then screened for their ability to interact with and modulate that predefined target [73].
Visualizing the Core Workflows
The fundamental difference between these approaches is encapsulated in their respective workflows, as illustrated below.
Comprehensive Comparative Analysis
Advantages and Disadvantages
The choice between PDD and TDD involves balancing distinct advantages and confronting specific challenges, as summarized in the table below.
Table 1: Advantages and Disadvantages of PDD and TDD
| Aspect | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| Key Advantage | Unbiased discovery of novel targets & mechanisms of action (MoA) [2] [11]. | Mechanistic clarity from the outset; rational, structure-based design is feasible [73]. |
| Target Space | Expands "druggable" space to include complex cellular machines & non-enzymatic targets [2] [9]. | Limited to known, predefined targets, often enzymes and receptors [11]. |
| Physiological Relevance | Higher; captures complex biology & polypharmacology in physiologically relevant models [2] [11]. | Lower; assays can be reductionist and may not capture full disease physiology [73]. |
| Clinical Translation | Historically higher rate of first-in-class drug discovery [2] [9]. | Can struggle with clinical translation despite promising target data [11]. |
| Primary Challenge | Target deconvolution can be difficult, time-consuming, and sometimes unsuccessful [11]. | Poor clinical translation if the target is not critically causal in the human disease [11] [73]. |
| Throughput & Cost | Can be lower throughput and more costly, especially with complex models [11]. | Generally higher throughput and more cost-effective for initial screening [73]. |
| Specificity | Risk of off-target effects harder to predict early on [11]. | High specificity for the intended target is a primary design goal [11]. |
Ideal Use Cases and Recent Successes
The strengths and weaknesses of each approach make them uniquely suited for different scenarios in the pipeline.
Table 2: Ideal Use Cases and Representative Drugs
| Application Scenario | Phenotypic Drug Discovery (PDD) | Target-Based Drug Discovery (TDD) |
|---|---|---|
| Ideal Use Cases | - Diseases with unknown or complex molecular etiology [11].- Discovering first-in-class medicines with novel MoAs [2].- Projects where polypharmacology (multi-target effect) is desirable [2].- When robust, disease-relevant phenotypic models exist. | - When a target is genetically validated with a strong link to disease [73].- Developing best-in-class drugs that improve on an existing MoA.- Rational drug design is possible with structural data (e.g., crystallography).- High-throughput screening of large chemical libraries is required. |
| Representative Approved Drugs | - Risdiplam (Spinal Muscular Atrophy): Modifies SMN2 splicing [2] [9].- Ivacaftor/Lumacaftor (Cystic Fibrosis): CFTR correctors/potentiators [2] [9].- Daclatasvir (Hepatitis C): NS5A inhibitor discovered via replicon screen [2] [9].- Vamorolone (Duchenne Muscular Dystrophy): Dissociative steroid [9]. | - Pimavanserin (Parkinson's disease psychosis): 5HT2a receptor inverse agonist [73].- Alirocumab/Evolucumab (High Cholesterol): PCSK9 inhibitors [73].- Imatinib (CML): BCR-ABL kinase inhibitor (though it exhibits polypharmacology) [2]. |
Detailed Experimental Protocols
A Representative PDD Protocol: High-Content Screening in a 3D Organoid Model
This protocol outlines a modern phenotypic screening campaign using a patient-derived colon cancer organoid model to identify compounds that reduce tumor growth.
1. Biological Model Selection and Validation:
- Material: Obtain patient-derived colon cancer organoids or generate from a validated cancer cell line (e.g., HT-29) in a basement membrane extract matrix [11].
- Validation: Characterize organoids via immunohistochemistry for key cancer markers (e.g., Ki-67, CD44). Ensure the model recapitulates key disease phenotypes like uncontrolled proliferation and resistance to apoptosis.
2. Assay Development and High-Content Imaging:
- Staining: At assay endpoint, fix organoids and stain with:
- Hoechst 33342: Nuclear stain for viability and count.
- Phalloidin-Alexa Fluor 488: Labels F-actin to visualize 3D cytoarchitecture.
- Cleaved Caspase-3 antibody (Alexa Fluor 594): Marker for apoptosis.
- Propidium Iodide: Marker for cell death.
- Imaging: Use a high-content confocal imager (e.g., PerkinElmer Operetta CLS or equivalent) to capture z-stacks of the entire organoid.
- Phenotypic Readouts: Quantify organoid size, volume, intensity of apoptosis/death markers, and morphological disruption.
3. Compound Screening and Hit Identification:
- Library: Screen a diverse, non-annotated chemical library (~10,000-100,000 compounds) [11].
- Execution: Dispense organoids into 384-well plates, treat with compounds (e.g., 10 µM final concentration), and incubate for 72-96 hours. Include controls (DMSO vehicle, reference cytotoxin).
- Analysis: Use AI-powered image analysis software (e.g., Ardigen's phenAID or similar) to extract hundreds of morphological features [9]. Apply machine learning models to identify "hit" compounds that induce a phenotype matching the desired profile (e.g., reduced size, increased apoptosis).
4. Counter-Screening and Lead Optimization:
- Specificity: Counter-screen hits for general cytotoxicity in non-malignant human fibroblast cells (e.g., WI-38) to exclude non-specific poisons.
- SAR: Perform medicinal chemistry to establish Structure-Activity Relationships (SAR) and improve the potency and properties of the lead series.
5. Target Deconvolution:
- Methods: Employ one or more of the following:
- Affinity Purification Mass Spectrometry: Immobilize the lead compound and pull down interacting proteins from cell lysates.
- Cellular Thermal Shift Assay (CETSA): Monitor target engagement by measuring protein thermal stability shifts upon compound binding.
- Functional Genomics: Use a CRISPR-Cas9 knockout or RNAi screen to identify genes whose loss of function mimics or rescues the compound-induced phenotype [11].
A Representative TDD Protocol: High-Throughput Screening for a Kinase Inhibitor
This protocol details a target-based screen to identify inhibitors for a specific kinase target, such as BRAF V600E, a known oncogenic driver.
1. Target Identification and Validation:
- Target: BRAF V600E kinase.
- Validation: Confirm the target's role via genetic (e.g., CRISPR knockout) and clinical evidence (e.g., mutation prevalence in melanoma).
2. Biochemical Assay Development:
- Assay Type: Homogeneous Time-Resolved Fluorescence (HTRF) kinase assay.
- Reagents:
- Recombinant BRAF V600E kinase protein.
- ATP and a specific peptide substrate.
- HTRF anti-phospho-substrate antibody conjugated with Europium cryptate.
- Streptavidin-conjugated XL665 to bind the biotinylated peptide.
- Principle: Kinase activity transfers a phosphate to the biotinylated substrate. The Europium cryptate antibody binds the phospho-substrate, and upon excitation, FRET occurs to the nearby XL665, producing a fluorescent signal at 665 nm. Inhibitors reduce this signal.
3. High-Throughput Screening (HTS):
- Execution: Run the assay in 1536-well plates. Dispense compound library (e.g., 500 nL of 1 mM stock), then add kinase, ATP, and substrate in a buffered solution. Incubate, followed by the addition of HTRF detection reagents.
- Readout: Measure the 665 nm/620 nm emission ratio on a plate reader (e.g., BMG Labtech PHERAstar).
- Hit Definition: Compounds showing >70% inhibition and a Z' factor >0.5 for the plate are considered primary hits.
4. Hit Validation and Selectivity Profiling:
- Dose-Response: Confirm hits in a 10-point dose-response curve to determine IC50 values.
- Selectivity Panel: Screen confirmed hits against a panel of 50-100 additional kinases to identify selective versus promiscuous inhibitors.
5. Cell-Based Assay and Lead Optimization:
- Cellular Model: Test compounds in a melanoma cell line (e.g., A375) harboring the BRAF V600E mutation.
- Mechanistic Readout: Measure inhibition of downstream signaling by Western blot (e.g., phospho-ERK levels).
- Reagents: Lysis buffer, primary antibodies (p-ERK, total ERK), HRP-conjugated secondary antibodies, ECL detection reagent.
- Optimization: Use structural data from co-crystallization of the lead compound with BRAF to guide medicinal chemistry for improved potency, selectivity, and drug-like properties (ADMET) [73].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for PDD and TDD
| Category | Item | Function & Application in Drug Discovery |
|---|---|---|
| Cellular Models | Patient-Derived Organoids | PDD: Provide physiologically relevant 3D models for phenotypic screening that mimic in vivo tissue architecture and disease states [11]. |
| iPSC-Derived Cells | PDD/TDD: Enable patient-specific disease modeling and screening; can be differentiated into relevant cell types (e.g., neurons, cardiomyocytes) [11]. | |
| Recombinant Cell Lines | TDD: Engineered to overexpress a specific molecular target for high-throughput target-based assays [73]. | |
| Assay Technologies | High-Content Imaging Reagents (e.g., fluorescent dyes, antibodies) | PDD: Enable multiplexed, quantitative analysis of complex phenotypic changes (morphology, signaling, viability) in cells and organoids [9] [11]. |
| HTRF/ALPHA Assay Kits | TDD: Provide robust, homogeneous, and miniaturized assay formats for high-throughput screening of enzymatic targets like kinases and GPCRs. | |
| Photoaffinity & Biophysical Probes (e.g., IMP-1575) | TDD/PDD (Deconvolution): Used for target identification and validation by covalently capturing drug-target interactions for proteomic analysis [73]. | |
| Compound Libraries | Diverse Small-Molecule Libraries | PDD/TDD: Foundation for screening campaigns; diversity is crucial for PDD to maximize chance of novel MoA discovery [11]. |
| Deconvolution Tools | CRISPR-Cas9 Knockout Libraries | PDD: Functional genomics tool for identifying genes essential for a compound's phenotypic effect, aiding in MoA elucidation [11]. |
| Affinity Purification Beads | PDD: Used to immobilize compounds and pull down bound proteins from complex biological lysates for target identification. |
The Integration of Artificial Intelligence
AI and machine learning are revolutionizing both PDD and TDD, acting as a force multiplier across the discovery pipeline [74] [75] [76].
- In PDD: AI-powered image analysis is critical for extracting rich, quantitative information from high-content screens. These tools can identify subtle phenotypic signatures and cluster compounds by their predicted MoA, significantly accelerating the target deconvolution process [9].
- In TDD: AI algorithms are used for de novo molecular design, generating novel drug-like structures optimized for a specific target. Tools like AlphaFold have dramatically improved protein structure prediction, enabling more accurate virtual screening and rational design [74] [76]. AI also predicts ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties early on, reducing late-stage failures [75] [77].
The synergy between modern PDD/TDD and AI is creating a new paradigm where data-driven insights compress discovery timelines, as evidenced by cases like Insilico Medicine's AI-designed drug candidate that advanced to preclinical trials in just 18 months [76].
The PDD versus TDD debate is not about declaring a single winner. The most successful drug discovery pipelines are those that strategically leverage the strengths of both approaches, often in an iterative manner. PDD excels at pioneering new therapeutic avenues and tackling biologically complex diseases, while TDD offers a streamlined path for optimizing interventions against validated targets.
The future of drug discovery lies in the intelligent integration of these approaches, powered by advanced disease models, functional genomics, and sophisticated AI tools. Researchers are encouraged to select their strategy based on the specific biological question at hand: choose PDD to explore the unknown and discover first-in-class therapies, and employ TDD to rationally engineer precision medicines against well-defined targets. By understanding the detailed advantages, limitations, and methodologies of each, drug development professionals can better navigate the path from concept to clinic.
Conclusion
The future of drug discovery does not lie in the strict adherence to a single paradigm but in the strategic integration of both phenotypic and target-based approaches. PDD has proven uniquely powerful for delivering first-in-class medicines with novel mechanisms of action, effectively expanding the druggable genome. Meanwhile, TDD remains a robust method for optimizing drug candidates against validated targets. The convergence of these strategies with cutting-edge technologies—especially AI, machine learning, and multi-omics integration—is creating a new, more powerful hybrid model. This synergistic framework leverages the unbiased, systems-level insight of PDD with the precision and rational design of TDD. For researchers, the path forward involves building adaptive workflows that select the best tool for the biological question at hand, thereby accelerating the development of safer and more effective therapies for complex human diseases.
References
- 1. Drug discovery [https://en.wikipedia.org/wiki/Drug_discovery]
- 2. Phenotypic Drug Discovery: Recent successes, lessons ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9708951/]
- 3. Phenotypic Screening - Advancing the science from ... [https://www.ddw-online.com/phenotypic-screening-advancing-the-science-from-discovery-to-regulatory-science-1119-201710/]
- 4. The Art of Finding the Right Drug Target: Emerging Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11334170/]
- 5. Discovery of RNA‐Targeting Small Molecules - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12375692/]
- 6. A review of transformer models in drug discovery and beyond [https://www.sciencedirect.com/science/article/pii/S2095177924001783]
- 7. Phenotypic Screening in Early Drug Discovery [https://proventainternational.com/phenotypic-screening-in-early-drug-discovery-opportunities-and-challenges/]
- 8. The future of phenotypic drug discovery [https://www.sciencedirect.com/science/article/pii/S2451945621000106]
- 9. Recent Successes in AI Phenotypic Drug Discovery and ... [https://ardigen.com/recent-successes-in-phenotypic-drug-discovery-and-the-future-of-ml-ai-methods/]
- 10. Phenotypic Drug Discovery Modern Successes [https://www.pfizer.com/news/articles/that_which_is_old_is_new_again_phenotypic_drug_discovery_makes_a_comeback]
- 11. Phenotypic Screening: A Powerful Tool for Drug Discovery [https://www.technologynetworks.com/drug-discovery/articles/phenotypic-screening-a-powerful-tool-for-drug-discovery-398572]
- 12. Phenotypic and targeted drug discovery in immune therapeutics [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra03914b]
- 13. Predicting compound activity from phenotypic profiles and ... [https://www.nature.com/articles/s41467-023-37570-1]
- 14. PhenoModel: A multimodal phenotypic drug design ... [https://www.sciencedirect.com/science/article/pii/S2211383525006446]
- 15. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 16. Recent advances in phenotypic drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7431967/]
- 17. Opportunities and challenges in phenotypic drug discovery [https://www.nature.com/articles/nrd.2017.111]
- 18. The future of phenotypic drug discovery [https://pubmed.ncbi.nlm.nih.gov/33529582/]
- 19. A Knowledge‐Guided Graph Learning Approach Bridging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12021103/]
- 20. Drug Discovery → Phenotypic-Based [https://drugdevelopment.web.unc.edu/drug-discovery-%E2%86%92-phenotypic-based/]
- 21. Assays for High Throughput Screening Drug Discovery [https://bellbrooklabs.com/high-throughput-screening-assays-drug-discovery/]
- 22. Target Identification & Validation in Drug Discovery [https://www.technologynetworks.com/drug-discovery/articles/target-identification-validation-in-drug-discovery-312290]
- 23. Target Validation [https://www.sygnaturediscovery.com/drug-discovery/integrated-drug-discovery/target-validation/]
- 24. Target Validation - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK195039/]
- 25. High-throughput screening [https://en.wikipedia.org/wiki/High-throughput_screening]
- 26. Adaptation of High-Throughput Screening in Drug Discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3269696/]
- 27. High Throughput Screening - an overview [https://www.sciencedirect.com/topics/chemistry/high-throughput-screening]
- 28. Phenotypic Drug Discovery: Models, Methodology and ... [https://www.frontiersin.org/research-topics/6115/phenotypic-drug-discovery-models-methodology-and-applications/magazine]
- 29. Phenotypic and targeted drug discovery in immune ... [https://pubs.rsc.org/en/content/articlelanding/2025/ra/d5ra03914b]
- 30. Machine Learning in Drug Discovery Market 2025 Trends [https://www.towardshealthcare.com/insights/machine-learning-in-drug-discovery-market-sizing]
- 31. Inside ELRIG's Drug Discovery 2025 [https://www.drugtargetreview.com/article/190237/inside-elrigs-drug-discovery-2025/]
- 32. New AI Tool Pinpoints Genes, Drug Combos To Restore ... [https://hms.harvard.edu/news/new-ai-tool-pinpoints-genes-drug-combos-restore-health-diseased-cells]
- 33. Top 10 AI Predictions for 2025 [https://aimagazine.com/articles/top-10-ai-predictions-for-2025]
- 34. The Future of Drug Discovery: Integrating Phenotypic Data ... [https://ardigen.com/the-future-of-drug-discovery-integrating-phenotypic-data-with-omics-and-ai/]
- 35. Advancing drug discovery through multiomics [https://www.ddw-online.com/advancing-drug-discovery-through-multiomics-37448-202510/]
- 36. Network-based multi-omics integrative analysis methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11954193/]
- 37. The latest trends in drug discovery [https://www.cas.org/resources/cas-insights/2025-drug-discovery-trends]
- 38. How AI and multiomics are accelerating drug discovery [https://www.ddw-online.com/how-ai-and-multiomics-are-accelerating-drug-discovery-38345-202510/]
- 39. Omics-based large language models: A new engine for ... [https://www.sciencedirect.com/science/article/pii/S2211383525007129]
- 40. CRISPR screening redefines therapeutic target ... [https://www.sciencedirect.com/science/article/pii/S2095177925001741]
- 41. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 42. High-content phenotypic and pathway profiling to advance ... [https://www.sciencedirect.com/science/article/pii/S2451945621001008]
- 43. Review Phenotypic screening with target identification and ... [https://www.sciencedirect.com/science/article/pii/S2451945621000969]
- 44. Why 90% of clinical drug development fails and how to ... [https://www.sciencedirect.com/science/article/pii/S2211383522000521]
- 45. Mitigating Trial Failure with Digital Adherence Monitoring [https://aardexgroup.com/mitigating-the-risk-of-trial-failure-with-digital-adherence-monitoring/]
- 46. Biopharma RD Faces Productivity And Attrition Challenges ... [https://www.clinicalleader.com/doc/biopharma-r-d-faces-productivity-and-attrition-challenges-in-2025-0001]
- 47. The unsustainability of clinical trials and need for change [https://www.clinicaltrialsarena.com/sponsored/the-unsustainability-of-clinical-trials-and-need-for-transformative-change/]
- 48. Target Identification vs. Phenotypic Screening in Drug ... [https://pharmafeatures.com/strategic-dichotomy-target-identification-vs-phenotypic-screening-in-drug-discovery/]
- 49. Phenotypic vs. target-based drug discovery for first-in-class ... [https://pubmed.ncbi.nlm.nih.gov/23511784/]
- 50. MultiVI: deep generative model for the integration of ... [https://www.nature.com/articles/s41592-023-01909-9]
- 51. Deep learning-based approaches for multi-omics data ... [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-024-00391-z]
- 52. A technical review of multi-omics data integration methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315550/]
- 53. Multimodal Integration in Health Care - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12370271/]
- 54. DRUG DISCOVERY - Overcoming Traditional Challenges [https://drug-dev.com/drug-discovery-overcoming-traditional-challenges-innovative-chemoproteomics-strategies-to-revolutionize-drug-discovery/]
- 55. Global first-in-class drugs approved in 2023–2024 [https://www.sciencedirect.com/science/article/pii/S2666675825000049]
- 56. Evidence-based support for phenotypic drug discovery in ... [https://www.sciencedirect.com/science/article/pii/S1359644622004007]
- 57. Dynamic clinical trial success rates for drugs in the 21st ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12572394/]
- 58. Dynamic clinical trial success rates for drugs in the 21st ... [https://www.nature.com/articles/s41467-025-64552-2]
- 59. Trends in FDA drug approvals: 2021–2024 insights & ... [https://www.sciencedirect.com/science/article/pii/S0003450925001063]
- 60. High Quality Hits in Drug Discovery: Confident Screening [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/high-quality-hits-in-drug-discovery/]
- 61. How AI is Transforming Drug Discovery Processes in 2025 [https://vasro.de/en/impact-ai-drug-discovery-2025/]
- 62. From Lab to Clinic: Success Stories of Repurposed Drugs in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535746/]
- 63. Unmodified methodologies in target discovery for small ... [https://www.sciencedirect.com/science/article/abs/pii/S1001841722003552]
- 64. AI-Driven Polypharmacology in Small-Molecule Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295758/]
- 65. Multi-target pharmacology: possibilities and limitations of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4585027/]
- 66. Polypharmacology: new drugs in 2023–2024 [https://link.springer.com/article/10.1007/s43440-025-00715-8]
- 67. Articles in 2025 | Nature Reviews Drug Discovery [https://www.nature.com/nrd/articles?year=2025]
- 68. Editorial: Multi-target drug discovery and design for ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1633600/full]
- 69. Target-based vs phenotypic drug discovery: opportunities and challenges with evidence-based application - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/B9780443239328000029]
- 70. 12 AI drug discovery companies you should know about [https://www.labiotech.eu/best-biotech/ai-drug-discovery-companies/]
- 71. BostonGene Named “AI-based Drug Discovery Solution of ... [https://bostongene.com/news-and-publications/news/bostongene-named-ai-based-drug-discovery-solution-of-the-year-in-2025-ai-breakthrough-awards-program]
- 72. 2025 AI in Drug Discovery: Predictions [https://lizard.bio/knowledge-hub/2025-ai-in-drug-discovery-predictions]
- 73. Promises and Challenges of Target-Based Drug Discovery [https://www.technologynetworks.com/drug-discovery/articles/promises-and-challenges-of-target-based-drug-discovery-355809]
- 74. AI in drug discovery: Use cases, market, and funding in 2025 [https://xenoss.io/blog/ai-drug-discovery]
- 75. AI-powered drug development: 2025 Revolution [https://lifebit.ai/blog/ai-powered-drug-development-2025-revolution/]
- 76. From Lab to Clinic: How Artificial Intelligence (AI) Is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298131/]
- 77. AI in Drug Discovery: Top Use Cases You Need To Know [https://smartdev.com/ai-use-cases-in-drug-discovery/]