Phenotypic vs Genotypic Resistance Testing: A Comprehensive Guide for Research and Drug Development
This article provides a comprehensive analysis of phenotypic and genotypic antimicrobial resistance testing methodologies for researchers and drug development professionals.
Phenotypic vs Genotypic Resistance Testing: A Comprehensive Guide for Research and Drug Development
Abstract
This article provides a comprehensive analysis of phenotypic and genotypic antimicrobial resistance testing methodologies for researchers and drug development professionals. It explores the fundamental principles distinguishing genetic potential from observable resistance, details current and emerging technological platforms, and offers comparative insights on performance validation. By synthesizing foundational concepts with practical applications, troubleshooting guidance, and comparative data, this resource aims to inform strategic decisions in antimicrobial development, surveillance, and diagnostic innovation.
Core Concepts: Defining Phenotypic and Genotypic Resistance
The genotype represents an organism's complete set of genetic material, comprising the specific alleles inherited from both parents that determine the potential for traits and biological functions [1] [2]. In contrast, the phenotype encompasses the observable characteristics—morphological, physiological, and behavioral—that result from the expression of this genetic information as influenced by environmental factors [3] [4]. This distinction, first formally proposed by Wilhelm Johannsen in 1911, remains fundamental to understanding heredity, evolution, and the mechanisms of disease [3] [4].
In antimicrobial resistance (AMR) research, this relationship takes on critical practical significance. Genotypic resistance refers to the presence of specific genetic determinants (e.g., mutations, acquired genes) within a pathogen's genome that confer the potential for resistance [5]. Phenotypic resistance describes the observable ability of a microbial population to survive or multiply despite antibiotic exposure, as determined through laboratory susceptibility testing [5]. The complex interplay between these concepts—where genetic potential does not always translate to observable expression—forms the cornerstone of modern resistance detection strategies and therapeutic decision-making.
Core Concepts and Definitions
Genotype: The Genetic Blueprint
The genotype constitutes the foundational genetic instruction set of an organism. Its composition and key characteristics are detailed below.
Table 1: Core Characteristics of Genotype
| Aspect | Description |
|---|---|
| Definition | The complete set of genetic material; the specific alleles an organism possesses [1] [2]. |
| Composition | DNA sequences, including protein-coding genes, regulatory sequences, and non-coding DNA [1] [6]. |
| Inheritance | Inherited from both parents, forming the genetic basis passed to offspring [1]. |
| Stability | Generally remains constant throughout life, barring mutations [1]. |
| Determination | Requires genetic testing methods (e.g., DNA sequencing, PCR, microarrays) [1] [2]. |
Genotypes are described using allelic combinations (e.g., AA, Aa, aa for a diploid organism) and can be homozygous (identical alleles) or heterozygous (different alleles) [6] [2]. The relationship between genotype and resultant phenotype is modulated by concepts such as penetrance (the proportion of individuals with a genotype who show the expected phenotype) and expressivity (the range of phenotypic severity among individuals with the same genotype) [6] [2].
Phenotype: The Observable Manifestation
The phenotype represents the physical and functional outcome arising from the interaction between the genotype and the environment.
Table 2: Core Characteristics of Phenotype
| Aspect | Description |
|---|---|
| Definition | The observable characteristics or traits of an organism, resulting from its genotype and environmental influences [1] [3]. |
| Composition | Physical attributes (morphology), biochemical properties, physiological processes, and behavior [1] [3]. |
| Inheritance | Not directly inherited; emerges from genotype-environment interaction [1]. |
| Stability | Can change dynamically due to environmental factors, nutrition, climate, and lifestyle [1] [6]. |
| Determination | Assessed through direct observation, measurement, or phenotypic assays [1] [5]. |
A key property of many phenotypes is phenotypic plasticity—the ability of a single genotype to produce different phenotypes in response to distinct environmental conditions [4] [7]. This plasticity is universal across life forms and can be a significant factor in evolution, potentially facilitating adaptation and the origin of novel traits [7].
Comparative Analysis: Genotypic Potential vs. Phenotypic Expression
The relationship between genotype and phenotype is not a simple one-to-one mapping. The following diagram illustrates the conceptual pathway and influencing factors from genetic potential to observable expression.
This complex interaction means that identical genotypes can yield different phenotypes (due to plasticity), and different genotypes can converge on similar phenotypes (due to genetic canalization or convergent evolution) [6] [4]. This has profound implications for predicting outcomes based on genetic information alone.
Application in Antimicrobial Resistance Testing
The genotype-phenotype distinction provides the conceptual framework for the two primary methodologies used in clinical and research laboratories to detect and characterize antimicrobial resistance.
Genotypic Resistance Testing
Genotypic testing identifies the genetic potential for resistance by detecting specific mutations or genes known to be associated with resistance mechanisms [5] [8].
Core Principle: The method involves identifying specific genetic markers—such as single-nucleotide polymorphisms (SNPs), insertions, deletions, or acquired genes (e.g., blaCTX-M, mecA)—within the pathogen's genome that are known to confer resistance [5] [9].
Protocol 1: Genotypic Resistance Testing via PCR and Sequencing
- Nucleic Acid Extraction: Isolate total DNA or RNA from a clinical sample (e.g., sputum, blood, urine) or a purified microbial culture. For RNA viruses, extract viral RNA.
- Amplification: For specific targets, use Polymerase Chain Reaction (PCR) with primers designed to flank the region of interest (e.g., a known resistance gene or mutation hotspot). For broader analysis, use whole-genome sequencing (WGS) approaches.
- Detection/Analysis:
- For Targeted PCR: Analyze amplification products via gel electrophoresis. The presence of a band of the expected size indicates the potential presence of the target gene.
- For Sequencing: Sequence the amplified PCR product or entire genome using next-generation sequencing platforms.
- Interpretation: Compare the generated DNA sequence to a wild-type (susceptible) reference sequence. Differences (mutations) are identified and interpreted using curated databases that link specific genetic changes to predicted resistance phenotypes [8].
Advantages and Limitations:
- Speed: Typically faster than phenotypic methods, yielding results in hours rather than days [9].
- Predictive Power: Can detect resistance potential even before it is expressed at a level detectable by phenotypic assays.
- Limitation: Only detects known mechanisms. A negative result does not rule out the presence of resistance conferred by novel, undetected genetic mechanisms [5]. Furthermore, the presence of a resistance gene does not always lead to its expression, potentially leading to overestimation of resistance.
Phenotypic Resistance Testing
Phenotypic testing directly measures the observable effect of an antimicrobial agent on microbial growth, providing a functional assessment of resistance [5] [10].
Core Principle: This approach exposes a standardized inoculum of the pathogen to a defined concentration of an antimicrobial drug and assesses the inhibition of growth, providing a direct measure of susceptibility [5] [9].
Protocol 2: Phenotypic Susceptibility Testing via Broth Microdilution
- Inoculum Preparation: Adjust the turbidity of a fresh, pure bacterial suspension to a standard density (e.g., 0.5 McFarland standard).
- Plate Preparation: Use a 96-well microtiter plate containing serial two-fold dilutions of various antibiotics in broth medium. This is often commercially prepared.
- Inoculation and Incubation: Dilute the standardized inoculum and add a precise volume to each well of the plate. Incubate under appropriate conditions (e.g., 35°C for 16-20 hours for many bacteria).
- Result Reading: Determine the Minimum Inhibitory Concentration (MIC)—the lowest concentration of antibiotic that completely prevents visible growth.
- Interpretation: Compare the MIC to established clinical breakpoints (e.g., from CLSI or EUCAST guidelines) to categorize the organism as Susceptible (S), Intermediate (I), or Resistant (R) [10].
Advantages and Limitations:
- Functional Readout: Directly measures the net effect of all resistance mechanisms present and expressed in the tested isolate, including novel mechanisms.
- Clinical Relevance: Results directly inform therapeutic decisions by indicating which drugs are effective.
- Limitation: The process is slower, typically requiring 18-24 hours or more after a pure culture is obtained [9]. It does not identify the specific genetic mechanism of resistance.
The following workflow summarizes the key steps and decision points in both genotypic and phenotypic testing methods.
Integrated Testing Approaches
Recognizing the complementary strengths and weaknesses of both methods, combined testing approaches are increasingly being adopted. For instance, the PhenoSense GT assay integrates genotypic and phenotypic data into a single report, providing information on both the key resistance mutations and the direct measurement of viral susceptibility to drugs [8]. This synergy offers clinicians the most comprehensive picture for designing effective treatment regimens.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of genotypic and phenotypic resistance studies requires a suite of specialized reagents and tools.
Table 3: Key Research Reagent Solutions for AMR Testing
| Item | Function/Application |
|---|---|
| PCR Master Mix | A pre-mixed solution containing Taq polymerase, dNTPs, buffers, and MgCl₂ for robust and specific amplification of target DNA sequences in genotypic assays [2]. |
| Broth Microdilution Panels | Pre-manufactured microtiter plates containing lyophilized or liquid serial dilutions of antibiotics. Essential for standardized, high-throughput phenotypic MIC determination [10]. |
| CLSI M100 / EUCAST Standards | Documents providing internationally recognized interpretive criteria (breakpoints) for MIC values and zone diameters, ensuring consistent reporting of Susceptible, Intermediate, and Resistant categories [10]. |
| Next-Generation Sequencing (NGS) Kits | Reagent kits for library preparation, target enrichment, and sequencing for comprehensive genotypic analysis, from single genes to whole genomes [2] [9]. |
| Quality Control Strains | Reference microbial strains with well-characterized genotypic and phenotypic resistance profiles. Used to validate the correct performance of both genotypic and phenotypic tests [10]. |
The precise distinction between genotypic potential and phenotypic expression is more than a theoretical concept; it is a practical framework that underpins modern approaches to combating antimicrobial resistance. Genotypic testing offers speed and predictive insight, while phenotypic testing provides a definitive functional assessment of resistance. The future of effective resistance management and personalized anti-infective therapy lies in leveraging the complementary nature of these two paradigms, often through integrated testing strategies. As technologies like whole-genome sequencing and artificial intelligence advance, the ability to more accurately predict phenotypic outcomes from genotypic data will continue to improve, further closing the loop between genetic potential and observable expression [9].
Antimicrobial resistance (AMR) represents one of the most significant challenges to modern healthcare, with nearly 700,000 annual global deaths attributed to resistant infections and projections estimating 10 million deaths annually by 2050 if current trends continue [11]. The rise of multidrug-resistant (MDR) pathogens, including Acinetobacter baumannii, Mycoplasma pneumoniae, and Escherichia coli, threatens to return modern medicine to a pre-antibiotic era where routine infections become untreatable [12] [13]. In this landscape, the speed and accuracy of antimicrobial resistance detection have transitioned from laboratory conveniences to critical determinants of patient survival and clinical outcomes.
The temporal relationship between appropriate antibiotic therapy and patient survival is unequivocal, particularly in bloodstream infections and sepsis, where delays in effective antimicrobial administration are directly associated with increased mortality [14]. Conventional antimicrobial susceptibility testing (AST) methods typically require 72 hours or more from specimen collection to results, forcing clinicians to rely on empiric therapy that may be ineffective against resistant strains [14]. This diagnostic delay creates a dangerous therapeutic gap during which patients may deteriorate despite broad-spectrum antibiotic coverage.
This application note examines the clinical imperative for rapid resistance detection technologies, framing the discussion within the broader context of phenotypic versus genotypic intrinsic resistance testing research. We present quantitative comparisons of current methodologies, detailed experimental protocols for implementation, and visualization of key pathways and workflows to guide researchers and clinicians in optimizing diagnostic strategies for improved patient outcomes.
Phenotypic vs. Genotypic Detection Methods: A Comparative Analysis
Fundamental Principles and Clinical Applications
Phenotypic and genotypic resistance detection methods operate on fundamentally different principles, each with distinct advantages and limitations in clinical practice. Phenotypic methods directly measure microbial growth or viability in the presence of antimicrobials, providing a functional assessment of resistance regardless of the underlying mechanism. These methods include disk diffusion, broth microdilution for minimum inhibitory concentration (MIC) determination, and novel rapid phenotypic technologies [14]. In contrast, genotypic methods detect specific genetic determinants of resistance, including resistance genes (e.g., bla-NDM, bla-OXA-48), point mutations (e.g., in gyrA, gyrB, parC), and gene overexpression mechanisms [12] [15] [11].
The clinical value of each approach depends heavily on context. Phenotypic methods remain the gold standard for therapy guidance as they directly measure the interaction between pathogen and drug [11]. However, genotypic methods offer significant speed advantages, potentially providing results within hours rather than days, which is crucial for timely therapeutic adjustments [12] [16]. Recent advances in rapid phenotypic technologies are bridging this temporal gap, with numerous platforms in development that promise phenotypic results in significantly reduced timeframes [14].
Quantitative Comparison of Method Performance
Table 1: Comparative Performance of Phenotypic and Genotypic Detection Methods for Key Pathogens
| Pathogen | Phenotypic Detection Range | Genotypic Detection Rate | Key Resistance Mechanisms | Clinical Impact |
|---|---|---|---|---|
| Acinetobacter baumannii (n=104) | 36.54%-89.42% [12] | 60% (56/93 isolates) [12] | OXA-48, NDM, VIM genes [12] | Life-threatening infections in critically ill patients [12] |
| Mycoplasma pneumoniae (n=121) | Not assessed | 47.9% macrolide resistance mutations [16] [13] | 23S rRNA gene mutations [13] | 2.20x higher macrolide use (aOR); 0.41x lower tetracycline use (aOR) with rapid testing [16] |
| Pasteurella multocida (n=80) | High susceptibility to cephalosporins, phenicols (>90%); high resistance to clindamycin, sulfamethoxazole [11] | strA, sul2, tetH most prevalent; no bla-TEM or erm(42) [11] | SNPs in gyrA (Ser83Ile, Ser83Arg, Asp87Asn) [11] | Strong correlation for phenicols, tetracyclines, fluoroquinolones; unexplained resistance to sulfamethoxazole, β-lactams [11] |
| Escherichia coli | Hypersensitivity to trimethoprim in knockout strains (ΔacrB, ΔrfaG, ΔlpxM) [15] | Efflux pump (acrB) and cell envelope (rfaG, lpxM) genes identified [15] | Efflux pumps, cell envelope biogenesis, LPS synthesis [15] | Targeting intrinsic resistance pathways sensitizes bacteria and limits resistance evolution [15] |
Correlation Between Methodologies
The relationship between phenotypic and genotypic resistance profiles varies significantly across pathogen-antibiotic combinations. For Pasteurella multocida, phenotypic results for phenicols, tetracyclines, and fluoroquinolones showed strong correlation with detected resistance genes, while resistance to sulfamethoxazole, β-lactams, and macrolides remained genetically unexplained, suggesting unidentified resistance mechanisms [11]. Similarly, in Acinetobacter baumannii, phenotypic methods demonstrated wider detection ranges (36.54%-89.42%) compared to genotypic methods (60%), indicating either superior sensitivity of phenotypic methods or limitations in current genetic target panels [12].
The broth microdilution method for MIC determination consistently shows stronger correlation with genotypic results compared to disk diffusion, making it more reliable for susceptibility testing despite being more resource-intensive [11]. This enhanced correlation likely reflects the quantitative nature of MIC data compared to the qualitative zone diameter measurements of disk diffusion.
The Impact of Rapid Testing on Clinical Decision-Making
Therapeutic Optimization and Antimicrobial Stewardship
Rapid resistance detection technologies directly influence antimicrobial prescribing patterns and stewardship outcomes. A retrospective study of 298 pediatric patients with Mycoplasma pneumoniae demonstrated that implementation of the Smart Gene Myco point-of-care test, which detects macrolide-resistance gene mutations, was associated with a 2.20-fold higher appropriate macrolide use (adjusted odds ratio) and a 0.41-fold lower tetracycline use (aOR) [16] [13]. This represents a significant optimization of antimicrobial selection based on rapid genotypic results, ensuring patients receive effective therapy while avoiding unnecessary broad-spectrum antibiotics.
The clinical value of rapid testing extends beyond initial therapy selection to encompass de-escalation strategies. Rapid exclusion of resistance mechanisms enables clinicians to confidently narrow antibiotic spectra, reducing collateral damage to commensal microbiota and selective pressure for resistance development [14]. This is particularly valuable in critical care settings, where inappropriate initial antibiotic therapy increases mortality risk by 2- to 3-fold in patients with bloodstream infections [14].
Temporal Advantages in Clinical Workflows
Table 2: Turnaround Time Comparison of Antimicrobial Susceptibility Testing Methods
| Testing Method | Time from Specimen Collection to Result | Time from Pure Colony to Result | Regulatory Status | Key Advantages |
|---|---|---|---|---|
| Conventional Phenotypic | ≥72 hours [14] | 4-24 hours [14] | Gold standard | Comprehensive, hypothesis-free |
| Rapid Phenotypic Technologies | Significantly faster than conventional [14] | <4 hours [14] | 12 with FDA 510(k)+CE; 6 with CE only [14] | Functional assessment, faster results |
| Genotypic Methods | Hours (post-culture) [12] | 2-8 hours [12] | Varies by platform | Rapid results, high specificity |
| Point-of-Care Genotypic | Potentially same-day [16] | <2 hours [16] | Emerging technologies | Bedside testing, immediate intervention |
The temporal advantage of rapid testing methodologies becomes clinically significant when considering the relationship between appropriate antibiotic therapy and patient survival. Studies consistently demonstrate that each hour of delay in effective antimicrobial administration increases mortality in septic patients by 7-10% [14]. Next-generation rapid phenotypic AST technologies can reduce total turnaround time from specimen collection to results through a combination of innovations, including direct specimen testing, reduced incubation periods, and accelerated detection methods [14].
Experimental Protocols for Resistance Detection
Protocol 1: Phenotypic Metallo-β-Lactamase Detection in Acinetobacter baumannii
Principle: This protocol detects MBL production in A. baumannii using both double-disk synergy tests and the MBL-E test, followed by confirmation with genotypic methods [12].
Materials:
- Mueller-Hinton agar plates
- Antibiotic disks: imipenem, meropenem, EDTA-containing disks
- MBL-E test strips or solutions
- Bacterial isolates (pure culture)
- PCR reagents for OXA-48, NDM, VIM gene detection
Procedure:
- Prepare 0.5 McFarland standard bacterial suspensions from fresh pure colonies.
- Lawn the suspensions onto Mueller-Hinton agar plates and allow to dry.
- For double-disk synergy test: Place imipenem and meropenem disks 20mm from EDTA-containing disks. Incubate at 35°C for 16-18 hours.
- For MBL-E test: Apply test strips or solutions according to manufacturer instructions. Incubate and observe for enhancement zones.
- Interpretation: Positive result indicated by ≥5mm increase in zone diameter between carbapenem and EDTA disks, or positive color change/zone in MBL-E test.
- Confirm positive phenotypes with PCR for OXA-48, NDM, and VIM genes.
Technical Notes: Among 93 drug-resistant A. baumannii isolates, phenotypic methods showed a detection range of 36.54%-89.42% compared to 60% with molecular methods [12]. Store antibiotic disks at -20°C until use. Include positive and negative controls in each batch.
Protocol 2: Genotypic Macrolide Resistance Detection in Mycoplasma pneumoniae
Principle: This protocol utilizes the Smart Gene Myco point-of-care testing platform to detect 23S rRNA gene mutations conferring macrolide resistance in M. pneumoniae [16] [13].
Materials:
- Smart Gene Myco test platform and reagents
- Nasopharyngeal swabs or respiratory specimens
- Nucleic acid extraction kit
- Positive and negative control samples
- Microcentrifuge and vortex mixer
Procedure:
- Extract nucleic acids from clinical specimens according to manufacturer instructions.
- Prepare reaction mixtures containing extracted nucleic acids, amplification reagents, and detection probes.
- Load samples into the Smart Gene Myco platform and initiate testing protocol.
- The system automatically performs nucleic acid amplification and detection of resistance mutations.
- Interpretation: Results indicate presence or absence of macrolide-resistance mutations in 23S rRNA gene.
- Report results to clinicians within 2 hours of specimen receipt.
Technical Notes: In clinical implementation, this test detected macrolide resistance mutations in 47.9% of pediatric patients, enabling more targeted antibiotic selection [16]. Ensure proper specimen collection and transport to maintain nucleic acid integrity.
Protocol 3: WHONET and R Software for AMR Surveillance
Principle: This protocol outlines a standardized approach for antimicrobial resistance surveillance using WHONET and R software for data analysis and visualization [17].
Materials:
- WHONET software (v.25.04.25)
- BacLink software (v.25.04.25)
- R software (v.4.4.0) with RStudio
- Microbiology laboratory data in compatible format (.csv, .txt, or laboratory-specific formats)
Procedure:
- Data Extraction: Export laboratory data including isolate identification, specimen type, collection date, and antibiogram results.
- Data Import with BacLink: Use BacLink to transform laboratory-native file formats into WHONET-compatible format.
- WHONET Configuration: Set up laboratory configuration in WHONET, including organism codes, antibiotic panels, and interpretation criteria (e.g., EUCAST guidelines).
- Data Analysis in WHONET: Generate resistance reports stratified by time period, pathogen, specimen type, or ward location.
- Data Export to R: Export aggregated resistance data from WHONET for advanced statistical analysis and visualization in R.
- Trend Analysis in R: Use R scripts to perform regression analysis of resistance trends over time and generate publication-ready figures.
Technical Notes: This standardized workflow enables reproducible AMR trend analysis with minimal resources, particularly valuable in low-resource settings [17]. The software tools are freely available, reducing economic barriers to implementation.
Visualization of Key Concepts and Workflows
Diagnostic Pathway for Antimicrobial Resistance Testing
Diagram 1: Diagnostic Pathway for Antimicrobial Resistance Testing. This workflow illustrates the parallel phenotypic and genotypic testing pathways from specimen collection to therapeutic decision-making, highlighting critical timepoints that impact patient outcomes.
Intrinsic Resistance Pathways in Escherichia coli
Diagram 2: Intrinsic Resistance Pathways in Escherichia coli. This diagram illustrates key intrinsic resistance mechanisms in E. coli, including efflux pumps and membrane barriers, and demonstrates how targeting these pathways can sensitize bacteria to antibiotics and reduce treatment failure risk [15].
Research Reagent Solutions for Resistance Studies
Table 3: Essential Research Reagents for Antimicrobial Resistance Studies
| Reagent Category | Specific Examples | Application/Function | Technical Notes |
|---|---|---|---|
| Culture Media | Mueller-Hinton Agar, Blood Culture Bottles | Supports bacterial growth for phenotypic testing | Quality control essential for reproducible results [12] |
| Antibiotic Disks/Panels | EUCAST-approved disks, Broth microdilution panels | Determinination of susceptibility profiles (MIC, zone diameters) | Store according to manufacturer specifications [11] |
| Molecular Detection Kits | Smart Gene Myco, PCR reagents for resistance genes | Detection of specific resistance mechanisms | Enables rapid, targeted resistance detection [16] |
| Whole Genome Sequencing | Library prep kits, BV-BRC, CARD database | Comprehensive resistance gene identification | Identifies known and novel resistance determinants [11] |
| Software Tools | WHONET, R with specialized packages | AMR surveillance data management and analysis | Enables trend analysis and resistance pattern identification [17] |
| Efflux Pump Inhibitors | Chlorpromazine, Piperine, Verapamil | Study of efflux-mediated resistance mechanisms | Demonstrates proof-of-concept for resistance breaking [15] |
The clinical imperative for rapid antimicrobial resistance detection is unequivocally established, with demonstrated impacts on mortality, antimicrobial stewardship, and healthcare costs. The complementary strengths of phenotypic and genotypic methods create a powerful synergistic relationship when integrated into diagnostic pathways. Phenotypic methods provide functional assessment of resistance across all possible mechanisms, while genotypic methods offer rapid results for targeted therapeutic adjustments.
Future directions in resistance detection will likely focus on technologies that combine the comprehensive nature of phenotypic assessment with the speed of genotypic methods. Next-generation rapid phenotypic platforms currently in development promise to bridge this temporal gap, potentially revolutionizing antimicrobial stewardship programs [14]. Additionally, the strategic targeting of intrinsic resistance pathways, such as efflux pumps and membrane barrier systems, represents a promising approach for resensitizing resistant pathogens to existing antibiotics [15].
Implementation of the protocols and methodologies outlined in this application note requires multidisciplinary collaboration between clinical microbiologists, infectious disease physicians, data scientists, and antimicrobial stewardship teams. Through systematic adoption of rapid, accurate resistance detection technologies and surveillance systems, healthcare institutions can significantly impact patient outcomes while combating the global threat of antimicrobial resistance.
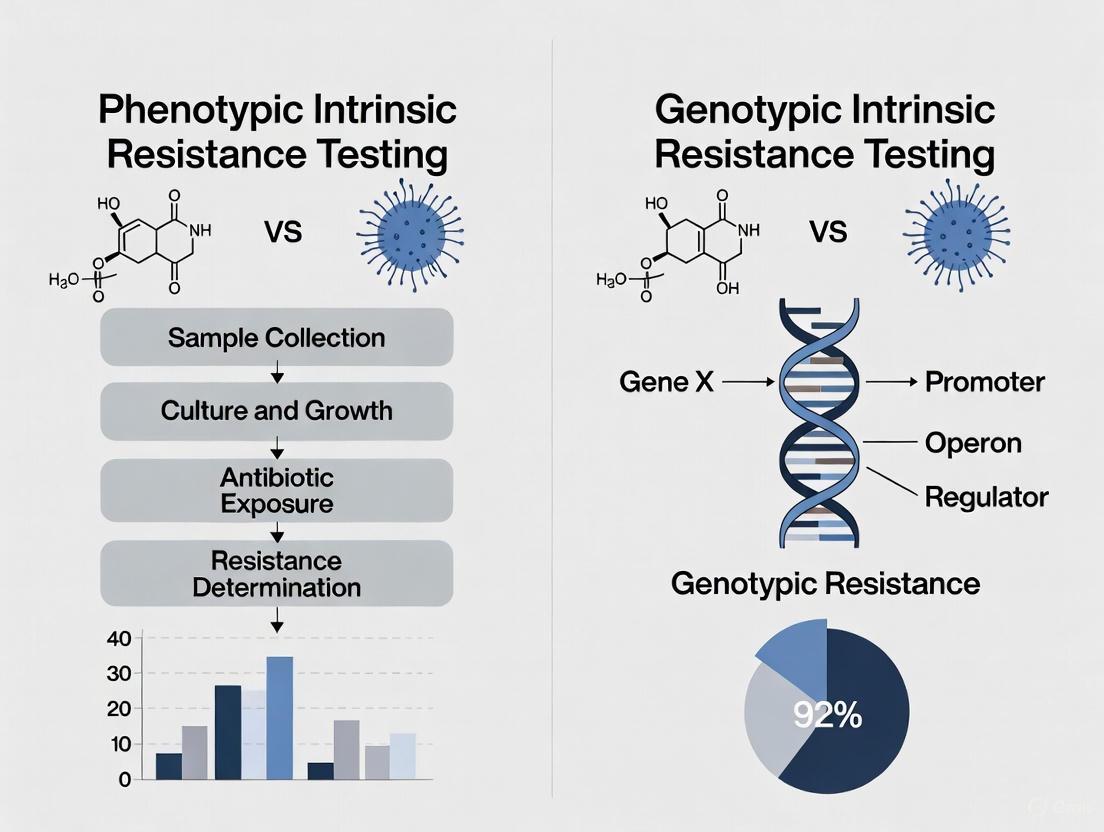
The escalation of antimicrobial and chemotherapeutic resistance represents a critical challenge in both clinical medicine and drug development. Resistance can be categorized into two fundamental concepts: genotypic resistance, which refers to the presence of specific genetic determinants that confer resistance potential (e.g., mutations in drug targets or acquired resistance genes), and phenotypic resistance, which describes the observable ability of a microbial or cancer cell population to survive and multiply despite therapeutic intervention [5]. These concepts are not mutually exclusive; rather, they represent different facets of the resistance spectrum. While genotypic testing reveals inherent resistance potential, phenotypic testing directly measures functional survival outcomes, providing the cornerstone for determining appropriate therapeutic strategies [5].
Understanding the mechanisms that bridge genetic mutations to functional survival is paramount for developing novel diagnostic tools and therapeutic interventions. This application note explores the quantitative methodologies and experimental protocols essential for delineating these mechanisms, providing researchers with structured frameworks to advance resistance testing in both microbiological and oncological research contexts within the broader scope of phenotypic versus genotypic intrinsic resistance investigations.
Quantitative Methodologies for Resistance Assessment
Minimum Inhibitory Concentration (MIC) Testing and Analysis
The minimum inhibitory concentration (MIC) test serves as a fundamental phenotypic assay for quantifying resistance levels in microbial isolates. The MIC defines the lowest concentration of an antimicrobial agent that inhibits visible growth of a microorganism [18]. These tests produce data points that fall within a range of concentrations rather than representing exact values, resulting in a data structure known as censoring. Proper analysis requires recognition of three censoring types: left-censoring (inhibition at all dilutions), right-censoring (no inhibition at highest concentration), and interval-censoring (inhibition between two concentrations) [18].
Table 1: Censoring Types in MIC Data Analysis
| Censoring Type | Description | Report Format |
|---|---|---|
| Left-Censored | Observation known only to be below the lowest concentration tested | ≤J μg/mL (where J is the lowest concentration) |
| Right-Censored | Observation known only to be above the highest concentration tested | >J μg/mL (where J is the highest concentration) |
| Interval-Censored | Observation known to lie between two concentration values | True MIC lies between reported MIC and one step below on two-fold scale |
For analytical purposes, MIC data can be categorized using established breakpoints. Epidemiological cutoff values (ECOFFs) separate wild-type (WT) isolates lacking acquired resistance mechanisms from non-wild-type (non-WT) organisms possessing detectable resistance mechanisms [18]. Alternatively, clinical breakpoints partition MIC values into susceptibility categories ("susceptible," "intermediate," and "resistant") based on clinical outcome data [18]. The choice between these categorization methods depends on the research objective, with ECOFFs更适合 for tracking resistance emergence and clinical breakpoints更适合 for predicting therapeutic efficacy.
Advanced Statistical Approaches for MIC Data
Analysis of MIC data requires specialized statistical approaches that account for censoring. Logistic regression models are widely employed but require dichotomization of the continuous MIC distribution, potentially losing information about shifts within susceptibility categories [18]. Cumulative logistic regression can accommodate multiple ordered categories (susceptible, intermediate, resistant) without presuming equal spacing between categories. For more sophisticated analyses, accelerated failure time-frailty models and mixture models preserve the interval-censored nature of MIC data, providing enhanced detection of subtle changes in MIC distributions that might be missed when data is categorized [18]. Model selection should consider the study objective, degree of censoring in the data, and consistency of testing parameters across experiments.
Experimental Protocols for Resistance Mechanism Investigation
Protocol for Assessing Resistance Development Risk to Microbicides
This protocol, adapted from research on microbicide resistance, provides a framework for evaluating the risk of resistance development following exposure to antimicrobial compounds [19]. The approach measures changes in microbicide and antibiotic susceptibility as primary markers for resistance emergence.
Table 2: Protocol for Microbicide Resistance Risk Assessment
| Step | Procedure | Key Parameters | Outcome Measures |
|---|---|---|---|
| 1. Strain Selection | Select target organisms including reference strains and industrial isolates | Include Gram-positive and Gram-negative species; restrict to maximum 2 subcultures from original stock | Diverse panel of test organisms |
| 2. Baseline Assessment | Determine pre-exposure MIC and MBC for formulations and active ingredients | Use doubling dilutions in culture medium; incubate 24h at 37°C | Baseline susceptibility profile |
| 3. Controlled Exposure | Expose strains to product formulations at concentrations yielding 1-3 log₁₀ reduction | Follow standardized suspension testing (e.g., BS EN 1276:2009); 1min exposure time | Surviving population for further testing |
| 4. Post-Exposure Assessment | Determine MIC and MBC of formulations and active ingredients after exposure | Same methodology as baseline assessment | Post-exposure susceptibility profile |
| 5. Antibiotic Susceptibility Profiling | Test baseline and post-exposure populations against panel of clinically relevant antibiotics | Kirby-Bauer disc diffusion or MIC determination | Changes in antibiotic susceptibility patterns |
| 6. Phenotype Stability Testing | Passage exposed populations without selective pressure | 5-10 passages in non-selective medium; reassay susceptibility | Stability of resistance phenotype |
This integrated protocol generates reproducible data for initial prediction of resistance development risk, employing cost-effective, high-throughput techniques that allow manufacturers to efficiently provide regulatory bodies with safety assessment data [19]. The methodology can be adapted for various antimicrobial agents beyond microbicides.
Dose-Escalation Protocol for Studying Chemotherapy Resistance
Investigations into irinotecan resistance in cancer cells demonstrate the importance of dose-escalation protocols for studying resistance evolution. The following methodology outlines key steps for observing adaptive resistance development:
Cell Line and Culture Conditions:
- Use cloned cancer cell lines (e.g., HCT116 colon cancer cells) to ensure genetic uniformity
- Isolate multiple independent single-cell clones (SCCs) to account for clonal variation
- For irinotecan studies, use activated derivative SN-38 due to inefficient activation in cell culture [20]
Dose-Escalation Treatment Scheme:
- Begin with initial exposure at IC₅₀ concentration (e.g., 4nM SN-38 for sensitive clones)
- Monitor cells for death and senescence-like growth arrest (typically 14 days initially)
- Resume growth when cells recover from arrest
- Repeat treatment cycle with same concentration (observe shorter arrest periods with each cycle)
- After adaptation to initial concentration, escalate dose (e.g., to 40nM) and repeat process
- Continue through multiple cycles (3-5) until cells demonstrate stable resistance [20]
Phenotypic Monitoring:
- Document duration of growth arrest following each exposure
- Quantify fraction of cell death after each treatment
- Assess morphological changes (enlargement, vacuolization indicative of senescence)
- Use cell cycle reporters to track arrest and re-entry into cell cycle [20]
Barcoding for Clonal Tracking:
- Implement cellular barcoding (e.g., Cellecta 50M barcodes lentiviral library) prior to selection
- Compare barcode frequencies pre- and post-selection to quantify clonal survival
- Assess difference between dose-escalation versus direct high-dose selection [20]
This approach demonstrates that dose escalation significantly enhances resistant variant development compared to direct high-dose exposure, with barcode analysis revealing 100-fold higher survival with escalation protocols [20].
Research Reagent Solutions
Table 3: Essential Research Reagents for Resistance Mechanism Studies
| Reagent/Cell Line | Specification | Research Application |
|---|---|---|
| HCT116 Colon Cancer Cells | Single-cell cloned populations | Irinotecan resistance studies; ensure genetic uniformity for adaptation experiments [20] |
| Mycobacterium tuberculosis Strains | Multiple lineage representatives (Lineages 1-4) | GWAS of drug resistance mechanisms; lineage-specific resistance propensity [21] |
| Cellecta 50M Barcodes Lentiviral Library | Complexity: 50 million unique barcodes | Clonal tracking in resistance evolution studies; quantifies population dynamics [20] |
| Salmonella enterica serovar Typhimurium | Strains SL1344 and 14028S | Microbicide resistance profiling; Gram-negative model for susceptibility testing [19] |
| Cell Cycle Reporter Systems | Fluorescent cell cycle indicators | Monitoring growth arrest and recovery in therapy resistance; distinguishes senescence from proliferation [20] |
| Whole Genome Sequencing Kits | Short-read and long-read technologies | Identification of resistance mutations; plasmid-encoded resistance gene detection [22] |
Signaling Pathways and Resistance Mechanisms
DNA Repair-Mediated Resistance in Chemotherapy
This diagram illustrates the mechanism of resistance development to Top1 inhibitors like irinotecan. Cancer cells initially sensitive to Top1 inhibitors due to increased cleavage sites gradually develop resistance through homology-directed repair of double-strand breaks, which reverts cleavage-sensitive "cancer" sequences back to cleavage-resistant "normal" sequences [20]. These mutations reduce DNA break generation upon subsequent exposures, leading to progressively increased drug resistance through dose escalation protocols.
DNA Repair Pathway Mutations in Antimicrobial Resistance
Genome-wide association studies of Mycobacterium tuberculosis clinical isolates have identified novel mutations in DNA repair genes (MutY, UvrA, UvrB, RecF) strongly associated with multidrug-resistant phenotypes [21]. These mutations collectively compromise DNA repair functions but paradoxically contribute to better bacterial survival under antibiotic and host stress conditions, facilitating the evolution of drug resistance through enhanced mutation rates or alternative survival pathways.
The intricate relationship between genetic mutations and functional survival outcomes underscores the complexity of resistance mechanisms across biological systems. The experimental frameworks and quantitative methodologies presented in this application note provide researchers with robust tools for dissecting these mechanisms, bridging the gap between genotypic prediction and phenotypic expression. As resistance continues to evolve, integrating these approaches within a comprehensive research strategy will be essential for developing next-generation diagnostics and therapeutics capable of overcoming adaptive survival mechanisms in both infectious diseases and oncology.
Within the framework of phenotypic versus genotypic intrinsic resistance research, a meticulously validated testing workflow is paramount. This protocol details the integrated application of phenotypic and genotypic methods to characterize intrinsic and acquired resistance mechanisms in bacteria. The strategic combination of these approaches provides a comprehensive resistance profile, bridging the gap between observable resistance phenotypes and their underlying genetic determinants [23] [22]. This is particularly critical for pathogens like Mycobacterium tuberculosis and multidrug-resistant Enterobacterales, where delayed or inaccurate susceptibility results can directly impact patient outcomes and facilitate the spread of resistance [23] [24]. The following sections provide a detailed, actionable pathway from specimen collection to final analysis, enabling researchers to obtain reliable, actionable data for informing therapeutic strategies and surveillance programs.
Research Reagent Solutions
The following toolkit comprises essential reagents and kits for executing the resistance testing workflow.
Table 1: Essential Research Reagents and Kits
| Item Name | Function/Application | Key Characteristics |
|---|---|---|
| Löwenstein–Jensen (L-J) Medium [23] | Solid culture medium for phenotypic DST of M. tuberculosis | Requires 28-35 days for results; considered a reference standard |
| BACTEC MGIT 960 System [23] | Automated liquid culture for rapid phenotypic DST | Average time to result: 7.5 ± 1.8 days |
| GenoType MTBDRplus Assay [23] | Line Probe Assay for genotypic detection of RIF and INH resistance | Detects mutations in rpoB (RIF) and katG/inhA (INH) |
| Anaerobic Transport Medium (ATM) [25] | Preserves viability of obligate anaerobes during transport | Specially designed to exclude oxygen; critical for abscess aspirates |
| Copan eSwab [25] | Collection swab for aerobic culture | Used for MRSA surveillance; non-inhibitory to anaerobic bacteria |
| DeepChek Assay & Software [26] | Targeted amplicon sequencing and bioinformatic analysis | Compatible with multiple NGS platforms for resistance mutation detection |
Methods
Specimen Collection and Transport
Proper specimen collection is the critical first step that determines the validity of all subsequent results.
- Site Selection and Sampling: Collect specimens from the primary site of infection, avoiding contamination with normal flora where possible [25]. For anaerobic cultures (e.g., from deep abscesses), obtain tissue biopsies or needle aspirates. Swabs are not acceptable for anaerobic culture due to poor recovery and potential for contamination [25].
- Choice of Collection Device:
- Transport: Label all specimens accurately. Transport to the laboratory promptly at room temperature (for anaerobic specimens) to preserve bacterial viability. Specimens in ATM should be processed within 24 hours of collection [25].
Phenotypic Drug Susceptibility Testing (DST)
This method determines resistance by observing bacterial growth in the presence of antibiotics.
Table 2: Phenotypic Drug Susceptibility Testing Methods
| Method | Principle | Time to Result | Key Applications |
|---|---|---|---|
| Löwenstein-Jensen (L-J) Solid Medium [23] | Growth of M. tuberculosis on antibiotic-containing solid medium | 28-35 days | Gold standard for first- and second-line TB DST |
| Liquid Culture (e.g., BACTEC MGIT 960) [23] | Automated detection of CO~2~ production by growing mycobacteria in liquid medium with antibiotics | ~7.5 days | Rapid phenotypic DST for M. tuberculosis |
| Broth Microdilution (MIC/MBC) [19] | Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) in a 96-well plate | 24 hours | Quantifying resistance levels; assessing cross-resistance risk |
Detailed Protocol: Broth Microdilution for MIC/MBC [19]
- Inoculum Preparation: Harvest an overnight broth culture by centrifugation. Resuspend the bacterial pellet in deionized water and standardize the suspension to 1 × 10^8^ CFU/mL.
- Plate Preparation: Prepare doubling dilutions of the antibiotic or microbicide of interest in a 96-well microtiter plate containing growth broth (e.g., Tryptone Soya Broth).
- Inoculation and Incubation: Add a standardized volume (e.g., 50 µL) of the bacterial inoculum to each well. Incub the plate at 37°C for 24 hours.
- MIC Determination: The Minimum Inhibitory Concentration (MIC) is defined as the lowest concentration of antimicrobial agent that completely inhibits visible growth.
- MBC Determination: Subculture broth from wells showing no growth onto antibiotic-free solid agar medium. The Minimum Bactericidal Concentration (MBC) is the lowest concentration that results in a ≥99.9% reduction in the original bacterial inoculum.
Genotypic Drug Resistance Detection
These methods identify resistance by detecting specific genetic mutations known to confer resistance.
3.3.1 Line Probe Assay (e.g., GenoType MTBDRplus) [23]
- DNA Extraction: Extract genomic DNA from a pure bacterial culture or directly from a clinical specimen.
- Amplification: Perform a multiplex PCR using biotinylated primers to amplify resistance-associated regions (e.g., rpoB for RIF, katG and inhA for INH).
- Hybridization and Detection: Denature the PCR products and hybridize them to membrane-bound probes. Subsequent enzymatic reaction produces a visible banding pattern.
- Interpretation: The presence or absence of specific wild-type and mutation bands determines the genotypic resistance profile.
3.3.2 Next-Generation Sequencing (NGS) for Resistance Profiling [26]
- Nucleic Acid Extraction: Extract high-quality genomic DNA or RNA from the sample.
- Library Preparation:
- Amplification: Use targeted primer sets (e.g., DeepChek Assays) to generate amplicons covering key drug resistance-associated genes.
- Fragmentation and Adapter Ligation: Fragment the amplicons enzymatically, then ligate platform-specific sequencing adapters.
- Quality Control: Verify library quality and quantity using a fragment analyzer (e.g., Agilent TapeStation) and fluorometry (e.g., Qubit).
- Sequencing: Load the library onto an NGS platform (e.g., Illumina ISeq100, Oxford Nanopore MinION). This protocol is compatible with multiple short- and long-read technologies [26].
- Bioinformatic Analysis: Process the raw sequencing data using specialized software (e.g., DeepChek). The workflow includes:
- Read trimming and alignment to a reference genome.
- Variant calling to identify mutations.
- Interpretation of mutations against a curated database of known resistance markers.
Diagram 1: Integrated resistance testing workflow showing parallel phenotypic and genotypic pathways.
Results and Data Interpretation
Concordance and Discordance Between Methods
The integration of phenotypic and genotypic data is where true actionable insight is generated.
Table 3: Example Concordance Between Genotypic and Phenotypic DST for M. tuberculosis (n=66 Isolates) [23]
| Drug | Phenotypic Resistance Rate (%) | Overall Concordance with Genotype MTBDRplus (%) | Notes on Discordance |
|---|---|---|---|
| Isoniazid (INH) | 84.85% | 95.16% | 2 cases: Genotypically susceptible but phenotypically resistant |
| Rifampicin (RIF) | 46.97% | 94.74% | 3 cases: Genotypically susceptible but phenotypically resistant |
| Streptomycin (STR) | 48.48% | Not specified | - |
| Ethambutol (EMB) | 30.30% | Not specified | - |
Interpreting Complex Results
- Actionable Result from Concordance: A genotype positive for a canonical rpoB mutation (e.g., S531L) and a corresponding resistant phenotype confirms MDR-TB, enabling immediate initiation of a second-line treatment regimen [23].
- Investigating Discordance: Isolates that are genotypically susceptible but phenotypically resistant (as noted in Table 3) suggest the presence of resistance mechanisms not detected by the genotypic assay (e.g., mutations in novel genes, efflux pump overexpression) [23] [24]. In such cases, the phenotypic result should take precedence for clinical decision-making, while further investigation via whole-genome sequencing is recommended for research.
- Beyond M. tuberculosis: The same principles apply to other pathogens. For example, in E. coli, intrinsic resistance mechanisms like efflux pumps (AcrB) and cell envelope integrity (LpxM, RfaG) are key regulators of susceptibility to diverse drug classes. Genotypic identification of such pathways can explain baseline phenotypic resistance and reveal targets for resistance-breaking adjuvants [24].
Diagram 2: Logic flow for integrating phenotypic and genotypic data to confirm a resistance mechanism.
This detailed protocol outlines a robust framework for moving from a clinical specimen to an actionable resistance profile. The synergistic use of phenotypic and genotypic methods compensates for the limitations inherent in each approach when used in isolation. Phenotypic testing provides a biologically relevant readout of resistance but is slow, while genotypic methods offer speed and precision but may miss novel or complex mechanisms [23] [27]. Adhering to this integrated workflow ensures that results are both timely and comprehensive, providing a solid foundation for effective patient management, antimicrobial stewardship, and ongoing resistance surveillance research.
Antimicrobial resistance (AMR) is a growing threat to global health, undermining the effectiveness of life-saving treatments and placing populations at heightened risk from common infections and routine medical interventions [28]. The World Health Organization's 2025 Global Antimicrobial Resistance and Use Surveillance System (GLASS) report provides a stark quantification of this burden, drawing on more than 23 million bacteriologically confirmed cases of bloodstream infections, urinary tract infections, gastrointestinal infections, and urogenital gonorrhoea from 110 countries [28]. This extensive data collection enables adjusted global and regional estimates of AMR for 93 infection type–pathogen–antibiotic combinations, providing researchers and public health officials with critical evidence to guide intervention strategies [28].
The clinical challenge is particularly acute with multidrug-resistant pathogens like Acinetobacter baumannii, a common cause of nosocomial infections. Recent studies reveal that 89.4% of A. baumannii isolates demonstrate drug resistance, creating life-threatening therapeutic challenges, especially in critically ill and vulnerable patients [12]. This resistance is conferred through various mechanisms, including metallo-beta-lactamase (MBL) production detected in up to 60% of isolates via molecular methods [12]. The relentless evolution of resistant pathogens has created an urgent need for innovative diagnostic approaches that can rapidly guide targeted antimicrobial therapy, preserving the efficacy of existing treatments while curbing unnecessary antimicrobial use.
The Diagnostic Innovation Imperative
The slow progress toward implementing conventional clinical bacteriology in low-resource settings, coupled with the universal need for greater speed in antimicrobial susceptibility testing (AST), has focused attention on next-generation rapid technologies [14]. Conventional AST workflows for bloodstream infections typically require a minimum of 72 hours from specimen collection to final susceptibility results, creating dangerous delays in appropriate antimicrobial administration [14]. This diagnostic gap fuels the overuse of empiric antimicrobials, further driving AMR emergence and spread.
The innovation imperative is particularly pressing in low-resource settings, where only approximately 1.3% of 50,000 medical laboratories in 14 sub-Saharan African countries offered any clinical bacteriology testing as of 2019 [14]. Barriers to conventional bacteriology implementation include requirements for specialized infrastructure, lack of automation, inadequate local access to complex supply chains, and human resource challenges [14]. Next-generation rapid phenotypic AST technologies promise to bridge this diagnostic gap by providing accurate susceptibility profiles within clinically relevant timeframes, potentially revolutionizing antimicrobial stewardship across diverse healthcare settings.
The Phenotypic vs. Genotypic Testing Paradigm
The debate between phenotypic and genotypic approaches for antimicrobial resistance detection represents a central frontier in diagnostic research. Phenotypic tests measure microbial growth or viability in the presence of antimicrobials to determine susceptibility, providing a functional assessment of resistance regardless of the underlying mechanism [14]. In contrast, genotypic methods detect specific resistance genes but may miss novel or unexpected resistance mechanisms.
Recent comparative studies highlight the complementary value of both approaches. A 2025 study comparing phenotypic and genotypic detection of drug resistance in Acinetobacter baumannii found that while molecular detection of drug-resistance conferring genes can be more time-effective, phenotypic methods provided valuable functional validation [12]. The researchers concluded that additional research is needed to develop comprehensive testing panels that integrate both approaches, as each provides distinct but complementary information about resistance profiles [12].
Table 1: Comparison of Phenotypic and Genotypic AMR Detection Methods
| Feature | Phenotypic Methods | Genotypic Methods |
|---|---|---|
| Basis of Detection | Microbial growth/viability in presence of antimicrobials | Detection of specific resistance genes |
| Time to Result | Traditionally 16-24 hours (conventional); newer rapid methods <8 hours | Typically 2-4 hours |
| Mechanism Coverage | Detects all resistance mechanisms regardless of genetic basis | Limited to known, targeted resistance genes |
| Clinical Correlation | Direct functional assessment | Indirect prediction based on gene presence |
| Example from Literature | MBL-E test, double-disk synergy test [12] | Molecular detection of OXA-48, NDM, VIM genes [12] |
Emerging Technologies in Rapid Phenotypic AST
A comprehensive review published in Nature Communications in 2024 synthesized the landscape of next-generation rapid phenotypic antimicrobial susceptibility testing, identifying over 90 distinct technologies at various development stages [14]. This analysis characterized technologies in terms of underlying technical innovations, technology readiness level (TRL), extent of clinical validation, and time-to-results from specimen collection. The review categorized technologies as commercialized (18 platforms, with 12 having FDA 510(k) clearance and CE marking) and non-commercialized (81 publications describing 67 phenotypic and 14 hypothesis-free nucleic acid-based platforms) [14].
These innovations employ diverse strategies to accelerate susceptibility testing, including:
- Direct specimen testing to eliminate culture steps
- Enhanced detection methods for early growth indication
- Microfluidic platforms for single-cell analysis
- Morphological changes detection using microscopy and AI
- Hypothesis-free nucleic acid-based tests using genomic recognition elements to detect or quantify bacteria in the presence of different antimicrobial conditions without pre-defined targets [14]
The standardized assessment of turnaround time from specimen collection revealed that many emerging technologies can provide reliable AST results within 8-24 hours compared to the conventional 72-hour workflow, representing a potentially practice-transforming advancement for clinical management of serious infections [14].
Case Study: Marple Rapid Diagnostic Tool
An exemplar of innovation targeting appropriate antimicrobial use is the Marple lateral flow device and smartphone app, currently in prototype phase and undergoing evaluation through a project led by Professor Jethro Herberg at Imperial College London with partners in The Gambia [29]. This test addresses the critical challenge of distinguishing between bacterial and viral respiratory infections using five immune response biomarkers identified through previous research [29].
Key features of this diagnostic tool include:
- Rapid results within 10 minutes
- Low production cost and minimal infrastructure requirements
- Smartphone app integration for algorithmic interpretation
- Pilot testing in both NHS and Gambian settings [29]
Professor Herberg emphasizes the potential impact: "A major driver of antimicrobial resistance is the unnecessary use of antibiotics in those who don't need them. By accurately finding out who has a bacterial infection and viral infection, we can ensure we only use antibiotics in those patients who need them" [29]. This project, funded by the £30 million PACE (Pathways to Antimicrobial Clinical Efficacy) initiative, represents the translation of 15 years of foundational research into a practical diagnostic solution with potential for global impact [29].
Table 2: Global AMR Surveillance Data and Response Initiatives
| Surveillance Metric | Reported Data | Significance |
|---|---|---|
| GLASS Data Volume | 23+ million bacteriologically confirmed cases [28] | Unprecedented scale of global AMR monitoring |
| Country Participation | 110 countries (2016-2023) [28] | Expanding global coordination in AMR surveillance |
| A. baumannii Drug Resistance | 89.4% of isolates (n=104) [12] | Highlights specific challenges with nosocomial pathogens |
| PACE Initiative Funding | £30 million [29] | Substantial investment in early-stage AMR innovation |
| EUP OHAMR Call Budget | €31+ million for 2026 [30] | Major transnational commitment to AMR research |
Experimental Protocols for AMR Detection
Protocol 1: Phenotypic Metallo-β-Lactamase Detection
Principle: This protocol detects metallo-β-lactamase (MBL) production in Gram-negative bacteria like Acinetobacter baumannii using a combination of double-disk synergy test, modified Hodge test, and MBL-E test [12].
Materials:
- Mueller-Hinton agar plates
- Imipenem and meropenem disks
- EDTA solution for MBL-E test
- Bacterial isolates (pure cultures)
- Incubator set at 35±2°C
Procedure:
- Prepare a 0.5 McFarland standard suspension of the test isolate in sterile saline.
- Lawn the suspension evenly onto Mueller-Hinton agar plates and allow to dry.
- For double-disk synergy test: Place an imipenem disk and EDTA-impregnated disk 15mm apart center-to-center.
- For MBL-E test: Use commercial MBL detection disks according to manufacturer instructions.
- Incubate plates at 35±2°C for 16-20 hours.
- Interpret results: Enhancement of the inhibition zone between the carbapenem and EDTA disks indicates MBL production.
Quality Control: Include known MBL-positive and MBL-negative control strains with each batch.
Protocol 2: Genotypic Detection of Carbapenem Resistance Genes
Principle: This protocol detects carbapenemase-encoding genes (OXA-48, NDM, VIM) in bacterial isolates using polymerase chain reaction (PCR) [12].
Materials:
- Bacterial DNA extraction kit
- PCR master mix
- Primers for OXA-48, NDM, and VIM genes
- Thermal cycler
- Gel electrophoresis system
- DNA molecular weight markers
Procedure:
- Extract genomic DNA from pure bacterial colonies using standardized protocols.
- Prepare PCR reaction mixtures containing:
- 12.5μL PCR master mix
- 1μL forward primer (10μM)
- 1μL reverse primer (10μM)
- 2μL DNA template
- 8.5μL nuclease-free water
- Run amplification with the following cycling conditions:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of: 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 1 minute
- Final extension: 72°C for 7 minutes
- Analyze PCR products by gel electrophoresis (1.5% agarose).
- Visualize under UV transillumination and document results.
Interpretation: Compare amplicon sizes with expected sizes for target genes (OXA-48: 744bp, NDM: 621bp, VIM: 390bp).
Research Reagent Solutions for AMR Detection
Table 3: Essential Research Reagents for AMR Detection Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Culture Media | Mueller-Hinton Agar, Blood Culture Bottles | Standardized growth conditions for phenotypic AST [12] [14] |
| Antimicrobial Disks | Imipenem, Meropenem, EDTA-impregnated disks | Disk diffusion assays for resistance phenotype detection [12] |
| Molecular Biology Kits | DNA Extraction Kits, PCR Master Mix, Primers | Genotypic detection of resistance genes (OXA-48, NDM, VIM) [12] |
| Reference Strains | ATCC control strains for MBL production | Quality control for assay validation [12] |
| Rapid Test Components | Lateral Flow Strips, Specific Antibodies | Development of rapid diagnostic devices (e.g., Marple test) [29] |
| Microfluidic Chips | Custom-designed single-cell analysis chips | Next-generation rapid phenotypic AST platforms [14] |
Workflow Diagrams for AMR Testing
Diagram 1: Conventional vs. Rapid AST Workflows. This diagram compares the sequential steps and time requirements for conventional AST versus emerging rapid technologies that integrate or parallelize testing approaches.
Diagram 2: Antimicrobial Resistance Mechanisms and Detection Methods. This diagram illustrates the major resistance mechanisms and their relationship to phenotypic and genotypic detection approaches.
The global AMR burden represents both a public health emergency and a powerful driving force for diagnostic innovation. The surveillance data from WHO GLASS and research on specific high-priority pathogens like Acinetobacter baumannii provide the evidentiary foundation for intensified efforts to develop, validate, and implement novel AST technologies [12] [28]. The landscape of over 90 rapid phenotypic AST technologies in development reflects a robust pipeline of innovation targeting the critical need for faster, more accessible susceptibility testing [14].
Future progress will require continued investment in translational research, as exemplified by the EUP OHAMR's forthcoming 2026 call "Treatments and adherence to treatment protocols" with a budget exceeding €31 million, involving 37 funding organizations from 28 countries [30]. This transnational collaboration will explore new combination treatments, tools to improve adherence to treatment protocols, and assessment of antimicrobial impacts across human, animal, and environmental health sectors [30]. As these innovations mature, the integration of phenotypic and genotypic approaches will likely yield comprehensive diagnostic solutions that preserve the efficacy of existing antimicrobials while curbing the further emergence and spread of resistance across the One Health spectrum.
Technological Platforms: From Conventional Methods to Next-Generation Tools
In the ongoing research on antimicrobial resistance (AMR), the debate between phenotypic and genotypic testing methods is central. While genotypic methods rapidly detect known resistance genes, phenotypic antimicrobial susceptibility testing (AST) remains the cornerstone for determining the actual response of bacteria to antimicrobial agents [31]. This document details the application and protocols for the three primary phenotypic gold standard methods, contextualized within intrinsic resistance research. These methods provide the definitive measurable phenotype against which genetic predictions must be ultimately correlated [32].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogues essential materials and their specific functions for implementing phenotypic AST methods, as derived from cited research.
Table 1: Essential Research Reagents and Materials for Phenotypic AST
| Item Name | Primary Function | Application Notes | Representative Examples (from search results) |
|---|---|---|---|
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized broth medium for dilution-based AST. | Ensures consistent ion concentration for reliable antibiotic activity. | Used in reference broth microdilution (BMD) methods [31]. |
| Iron-Depleted Mueller-Hinton (ID-MH) | Specialized medium for testing siderophore antibiotics. | Creates iron-limited conditions essential for drug uptake. | Critical for accurate cefiderocol testing [33]. |
| Commercial BMD Panels | Pre-configured microdilution plates for efficient MIC testing. | Validated for specific organism groups; reduces preparation time. | MICRONAUT-S Anaerobes MIC; Sensititre Anaerobe MIC [34]. |
| Gradient Diffusion Strips | Impregnated plastic strips for determining MIC on agar. | Provides flexibility for single-agent/single-isolate testing. | Liofilchem MIC Test Strips (MTS); bioMérieux Etest [34] [35]. |
| Antimicrobial Disks | Paper disks containing a defined antibiotic concentration for diffusion assays. | Enables qualitative susceptibility profiling and resistance screening. | Used in Kirby-Bauer disk diffusion (DD) method [33]. |
| Breakpoint Tables | Interpretive criteria defining S, I, and R categories. | Essential for translating MIC or zone diameter into a clinical prediction. | EUCAST Clinical Breakpoint Tables; CLSI M100 [36] [10]. |
Methodological Deep Dive: Protocols and Performance
Broth Microdilution (BMD)
Detailed Experimental Protocol:
- Preparation: Use sterile, cation-adjusted Mueller-Hinton broth (CAMHB) for most aerobes. For fastidious organisms or specific drugs (e.g., cefiderocol), use specialized media like ID-MH [33].
- Inoculation: Prepare a standardized bacterial suspension (e.g., 0.5 McFarland) in saline or broth. Dilute the suspension to achieve a final inoculum of approximately 5 × 10^5 CFU/mL in each well of the microdilution plate [31].
- Incubation: Seal the plate to prevent evaporation and incubate at 35±2°C for 16-20 hours under ambient air. Incubation for 48 hours may be required for slow-growing bacteria or anaerobes [34].
- Reading and Interpretation: Examine each well for turbidity. The Minimum Inhibitory Concentration (MIC) is the lowest concentration of antimicrobial that completely inhibits visible growth. Compare the MIC to FDA or EUCAST breakpoints for categorical interpretation (S, I, R) [36] [10] [37].
Gradient Diffusion Method (GDM)
Detailed Experimental Protocol:
- Inoculation: Evenly swab a standardized bacterial suspension (0.5 McFarland) onto the surface of a suitable agar plate (e.g., Mueller-Hinton agar) to create a confluent lawn.
- Strip Application: Using sterile forceps, carefully apply the gradient strip onto the agar surface, ensuring full contact.
- Incubation: Invert the plate and incubate at 35±2°C for 16-20 hours.
- Reading and Interpretation: After incubation, the ellipse of inhibition will be visible. The MIC is read at the point where the ellipse's edge intersects the strip's scale [35]. This value is then interpreted using clinical breakpoints.
Disk Diffusion (DD)
Detailed Experimental Protocol:
- Inoculation and Disk Application: As with GDM, prepare a confluent bacterial lawn on Mueller-Hinton agar. Apply antibiotic disks to the surface using a sterilized dispenser or forceps, ensuring adequate spacing to prevent overlapping zones.
- Incubation: Invert and incubate the plate at 35±2°C for 16-18 hours.
- Reading and Interpretation: Measure the diameter of the complete inhibition zone (including the disk diameter) to the nearest millimeter. Interpret the zone diameter using standardized tables (e.g., EUCAST, CLSI) to categorize the isolate as Susceptible, Intermediate, or Resistant [33].
Comparative Performance Data
Recent studies highlight the performance characteristics of these methods against reference standards and each other.
Table 2: Comparative Analytical Performance of Phenotypic AST Methods
| Method | Organism Group | Essential Agreement (EA) with Reference | Categorical Agreement (CA) with Reference | Key Error Notes |
|---|---|---|---|---|
| Broth Microdilution (Commercial Kits) | Clostridiales spp. [34] | Variable (48h incubation improved EA for some drugs) | >90% for metronidazole, piperacillin/tazobactam, vancomycin | Highest error rate for clindamycin |
| Gradient Diffusion Strips | Clostridiales spp. [34] | Lower than BMD for some drugs; acceptable for vancomycin | Above FDA threshold except for clindamycin & penicillin G | - |
| Gradient Diffusion (Etest) | Neisseria gonorrhoeae [35] | 95-96% with Agar Dilution | 66-83% for "alert" MICs (CRO, CFX, AZM) | Effective for rapid detection of emerging resistance |
| Commercial BMD (ComASP) | P. aeruginosa (Cefiderocol) [33] | 82.1% | 94.0% | Reliable for diagnostic use, possibly in combination |
| Commercial BMD (UMIC) | P. aeruginosa (Cefiderocol) [33] | 74.4% | 78.6% | Tendency to overestimate MICs |
Diagram 1: AST Workflow from Culture to Interpretation.
Critical Considerations for Research & Development
- Regulatory and Standards Alignment: A major update in 2025 saw the FDA fully recognize breakpoints from key CLSI standards (M100, M45), resolving a significant hurdle for test developers and ensuring U.S. laboratories can use current interpretive criteria [10] [37]. Researchers must align protocols with the latest EUCAST or CLSI guidelines.
- Resolving Genotype-Phenotype Discrepancies: Phenotypic AST is the definitive arbiter when genotypic predictions conflict with observed growth inhibition. A structured approach is required to investigate discrepancies, which may arise from off-target resistance mechanisms, gene expression levels, or technical factors [32].
- Tackling Technical Challenges: Specific organisms and antimicrobials present unique challenges. Testing anaerobes requires extended incubation (up to 48 hours) and specialized methods like the agar dilution reference [34]. Similarly, testing siderophore antibiotics like cefiderocol demands iron-depleted media to accurately simulate the in vivo environment and drug uptake [33].
Broth microdilution, disk diffusion, and gradient diffusion strips form an indispensable toolkit for grounding AMR research in measurable phenotypic reality. The choice of method depends on the required output (quantitative MIC vs. qualitative categorization), throughput, and organism-drug combination. As regulatory landscapes evolve and the complexity of resistance mechanisms increases, these phenotypic gold standards continue to provide the critical baseline against which novel genotypic methods and their predictions must be validated.
Application Notes
Next-generation phenotypic platforms represent a paradigm shift in biomedical research, moving beyond static, endpoint observations to dynamic, multi-parameter analyses of living systems. These integrated technological approaches—morphokinetic, microfluidic, and spectroscopic—provide unprecedented resolution for characterizing complex biological processes, from cellular development to drug resistance mechanisms. By capturing the temporal, spatial, and molecular dimensions of phenotypic expression, these platforms enable researchers to decipher the functional consequences of genetic variation and environmental perturbations with remarkable precision.
The integration of these technologies is particularly transformative for studying intrinsic resistance, where conventional genotypic approaches often fail to predict therapeutic outcomes due to the complex, multifactorial nature of drug resistance. Where genotypic testing identifies potential resistance markers based on genetic sequences, phenotypic platforms directly measure functional responses, capturing emergent properties that arise from complex biological networks. This capability is crucial for understanding non-genetic resistance mechanisms, adaptive cellular states, and the contribution of minority cell populations to treatment failure.
Technology-Specific Application Notes
Morphokinetic Profiling Platforms
Morphokinetic analysis leverages time-lapse monitoring to quantify the dynamic morphological changes and division kinetics of cells or embryos in response to experimental perturbations. This approach has demonstrated significant utility in predicting functional outcomes, including treatment efficacy and developmental potential.
In assisted reproduction research, morphokinetic parameters have proven highly predictive of live birth outcomes. A retrospective analysis of 429 blastocysts revealed that specific timing parameters, particularly the time to 5-blastomere stage (t5) and time to start of blastulation (tSB), showed statistically significant differences between embryos that resulted in live births versus those that failed to implant. Embryos with optimal morphokinetic parameters exhibited substantially higher developmental potential, enabling more reliable selection criteria [38].
The integration of artificial intelligence with morphokinetic analysis has further enhanced predictive accuracy. A comparative study evaluating 91 IVF cycles found that automated scoring systems (KIDScore and iDAScore) applied to time-lapse monitoring data effectively predicted live birth outcomes. KIDScore D5, which incorporates both morphokinetic parameters and morphological assessment, demonstrated particularly robust performance in identifying embryos with high implantation potential, providing a decision-support tool that outperforms conventional morphological assessment alone [39].
Table 1: Key Morphokinetic Parameters for Outcome Prediction
| Parameter | Biological Significance | Measurement Method | Predictive Value |
|---|---|---|---|
| t5 (time to 5 cells) | Embryo cleavage kinetics | Time-lapse imaging at 10-min intervals | Strong correlation with live birth outcome [38] |
| tSB (start of blastulation) | Initiation of differentiation process | Automated annotation of blastocoel formation | Significant predictor of implantation success [38] |
| KIDScore D5 | Composite algorithm of morphokinetics & morphology | AI-based analysis of time-lapse sequences | High efficiency in predicting live birth [39] |
| iDAScore | Fully automated blastocyst assessment | 3D convolutional neural network analysis | Correlates with reproductive outcome [39] |
Microfluidic Single-Cell Analysis Platforms
Microfluidic technologies enable high-resolution single-cell analysis under precisely controlled microenvironments, making them particularly valuable for detecting rare cell subpopulations that may contribute to intrinsic resistance. The single-cell encapsulation approach allows researchers to investigate cellular heterogeneity and identify resistant subsets that would be masked in bulk population analyses.
This technology operates on principles of pressure-driven flow and droplet generation, creating picoliter-scale aqueous compartments in an immiscible oil phase that serve as isolated microreactors for individual cells. The application of Poisson distribution statistics ensures optimal loading conditions, with recommended lambda values (λ) of 0.05-0.1 resulting in approximately 5% of droplets containing a single cell, while over 90% remain empty, and less than 0.5% contain multiple cells [40].
The applications of single-cell encapsulation span multiple research domains, each benefiting from the ability to resolve cellular heterogeneity:
- Oncology: Identifying rare resistant subpopulations carrying critical mutations
- Immunology: Characterizing diverse immune cell types and rare immune subsets
- Neurobiology: Constructing detailed neuronal maps and connection patterns
- Developmental Biology: Tracing lineage commitment and embryonic development
- Single-Cell Omics: Revealing tissue heterogeneity at unprecedented resolution [40]
This approach is particularly powerful for intrinsic resistance research, as it enables the functional characterization of rare persister cells that survive drug treatment through non-genetic adaptive mechanisms, potentially serving as reservoirs for eventual resistance development.
Spectroscopic Interaction Assays
Spectroscopic techniques provide label-free methods for quantifying molecular interactions in real-time, offering insights into the binding events that underlie drug efficacy and resistance mechanisms. These technologies have evolved toward increasingly sensitive detection limits, reduced sample requirements, and higher throughput capabilities.
Table 2: Comparative Analysis of Spectroscopic Interaction Platforms
| Technique | Detection Principle | Key Applications | Sensitivity | Advantages |
|---|---|---|---|---|
| Surface Plasmon Resonance (SPR) | Reflectivity changes from surface plasmon waves | Protein-protein interactions, drug-target binding | pmol/L | Real-time kinetics, industry gold standard [41] |
| Bio-Layer Interferometry (BLI) | White light interference pattern shifts | Protein-small molecule interactions, antibody characterization | pmol/L | Simple operation, minimal sample consumption [41] |
| Back-Scattering Interferometry (BSI) | Refractive index changes in free solution | Molecular interactions without immobilization | Not specified | Free-solution technique, no surface attachment needed [41] |
| Microscale Thermophoresis (MST) | Molecular movement in temperature gradients | Binding affinity in solution | Not specified | Solution-based measurement, minimal sample preparation [41] |
Recent applications demonstrate the utility of these platforms in resistance research. SPR technology has been employed to validate interactions between drug candidates and their protein targets, such as the characterization of NeoPrzewaquinone A binding to IL-15Rα, revealing high specificity and binding affinity that suggests potential therapeutic utility [41]. Similarly, BLI has been used to document interactions between traditional Chinese medicine compounds (quercetin, wogonin, rutin) and inflammatory targets (TNF-α, IL-6, IL-1β), providing mechanistic insights into their anti-inflammatory effects [41].
Experimental Protocols
Morphokinetic Embryo Assessment Protocol
Equipment and Reagents
- Time-Lapse Monitoring System: Embryoscope+ incubator (Vitrolife) with EmbryoViewer software (v7.8.2 or higher)
- Culture Media: Sage 1-Step single-step culture media (Origio) or equivalent
- Culture Dishes: EmbryoSlide+ specialized culture dish (Vitrolife)
- Oil Overlay: OVOIL mineral oil (Vitrolife)
- Gas Mixture: 5.0% O₂, 6.6% CO₂, balanced N₂
Step-by-Step Procedure
Oocyte Preparation and Fertilization:
- Perform oocyte retrieval 34-36 hours after ovulation triggering with 250μg choriogonadotropin alfa
- Denude cumulus cells and perform ICSI approximately 40 hours post-trigger
- Assess fertilization 16-18 hours post-ICSI by confirming two pronuclei (2PN)
Embryo Loading and Culture:
- Transfer normally fertilized oocytes to individual microwells of pre-equilibrated EmbryoSlide+
- Ensure complete coverage with OVOIL mineral oil to prevent evaporation
- Load dish into Embryoscope+ time-lapse incubator maintaining stable conditions (37°C, 5.0% O₂, 6.6% CO₂)
Image Acquisition and Morphokinetic Annotation:
- Acquire images at 10-minute intervals across 11 focal planes throughout 5-6 day culture period
- Annotate key developmental milestones using EmbryoViewer software:
- Time of division to 2-cells (t2), 3-cells (t3), 4-cells (t4), 5-cells (t5), 6-cells (t6), 7-cells (t7), 8-cells (t8)
- Time to compaction (tM)
- Time to start of blastulation (tSB)
- Time to full blastocyst (tB)
Automated Scoring Application:
- Apply KIDScore D5 algorithm for day 5 blastocysts, which generates a score from 1-9.9 based on known implantation data
- Apply iDAScore algorithm, which uses 3D convolutional neural network analysis of full time-lapse sequences
- Transfer or cryopreserve embryos with the highest combined scores for potential clinical use
Morphokinetic Analysis Workflow
Microfluidic Single-Cell Encapsulation Protocol
Equipment and Reagents
- Flow Controller: OB1 pressure controller with at least two 0-2000 mbar channels (Elveflow)
- Flow Sensors: MFS 2D (0.4-7 μL/min) for oil phase, MFS 3D (4.2-80 μL/min) for cell suspension
- Microfluidic Chip: PDMS chips with fluid-focusing geometry and hydrophobic channels (Microfluidic ChipShop)
- Oil Phase: HFE-7500 fluorinated oil with 1% FluoSurf surfactant (Emulseo)
- Surface Treatment: Aquapel (Autoserv) for hydrophobic channel treatment
Step-by-Step Procedure
System Setup and Priming:
- Connect OB1 pressure controller to computer via USB and launch control software
- Calibrate pressure channels and connect flow sensors according to manufacturer specifications
- Treat microfluidic chip channels with Aquapel for hydrophobicity:
- Flush chip with argon gas
- Inject Aquapel for surface treatment
- Flush again with argon gas
- Rinse with HFE-7500 oil phase
Solution Preparation:
- Prepare oil phase: HFE-7500 with 1% FluoSurf surfactant
- Prepare cell suspension at optimal concentration based on Poisson distribution:
- Calculate concentration for λ = 0.05-0.1
- For 50μm droplets: ~1.375×10⁶ cells/mL yields ~8.7% single-cell encapsulation
- Load solutions into 15mL Falcon tubes with appropriate tubing
Droplet Generation:
- Prime chip with oil phase at low pressure (50 mbar) until droplets form at outlet
- Connect cell suspension line and establish stable flow
- Set flow rates:
- Oil phase: 300 mbar pressure or 45 μL/min flow rate
- Cell suspension: 220 mbar pressure or 10 μL/min flow rate
- Adjust parameters to achieve desired droplet size (typically 50-80μm diameter)
Collection and Analysis:
- Collect emulsion in outlet reservoir containing HFE-7500 with 1% FluoSurf
- Monitor droplet quality and single-cell encapsulation rate
- For HeLa cells at recommended concentration, expect:
- 90.9% empty droplets (theoretical: 91.4%)
- 8.7% single-cell droplets (theoretical: 8.2%)
- 0.4% multi-cell droplets (theoretical: 0.4%)
Single-Cell Encapsulation Workflow
Surface Plasmon Resonance (SPR) Binding Protocol
Equipment and Reagents
- SPR Instrument: Commercial SPR system with microfluidic flow cells
- Sensor Chips: Appropriate surface chemistry (CM5 for carboxylated dextran)
- Running Buffer: HBS-EP (10mM HEPES, 150mM NaCl, 3mM EDTA, 0.005% surfactant P20, pH 7.4)
- Ligand and Analyte: Purified proteins or molecules of interest
Step-by-Step Procedure
Sensor Chip Surface Preparation:
- Dock appropriate sensor chip into instrument
- Prime system with running buffer until stable baseline achieved
- Activate carboxylated dextran surface with EDC/NHS chemistry (7 min, 10μL/min)
Ligand Immobilization:
- Dilute ligand in appropriate immobilization buffer (typically sodium acetate, pH 4.0-5.5)
- Inject ligand solution (5-10μg/mL) for 7 minutes at 10μL/min to achieve target immobilization level
- Block remaining activated groups with ethanolamine (7 min, 10μL/min)
- Establish stable baseline with running buffer flow
Binding Interaction Analysis:
- Prepare analyte serial dilutions in running buffer
- Program automated run method:
- Baseline: 1-2 minutes with running buffer
- Association: 3-5 minutes with analyte solution
- Dissection: 5-10 minutes with running buffer
- Regeneration: 30-60 seconds with regeneration solution (typically glycine pH 2.0-3.0)
- Inject each analyte concentration in duplicate or triplicate
Data Analysis and Interpretation:
- Subtract reference cell and blank injection responses
- Fit binding curves to appropriate interaction models (1:1 Langmuir binding for simple interactions)
- Calculate kinetic parameters:
- Association rate constant (kₐ)
- Dissociation rate constant (k_d)
- Equilibrium dissociation constant (KD = kd/kₐ)
The Scientist's Toolkit
Table 3: Essential Research Reagents and Solutions
| Item | Specifications | Application | Key Function |
|---|---|---|---|
| EmbryoSlide+ Culture Dish | Specialized microwell design for time-lapse imaging | Morphokinetic analysis | Individual embryo culture with minimal disturbance during imaging [39] |
| HFE-7500 + FluoSurf | Fluorinated oil with 1% fluorosurfactant | Microfluidic encapsulation | Forms stable, biocompatible emulsion for single-cell analysis [40] |
| CM5 Sensor Chip | Carboxylated dextran matrix on gold surface | SPR analysis | Provides versatile surface for ligand immobilization [41] |
| OB1 Pressure Controller | 0-2000 mbar pressure range, multiple channels | Microfluidics | Delivers precise, pulseless flow for stable droplet generation [40] |
| Sage 1-Step Media | Single-step culture medium formulation | Embryo culture | Maintains embryo viability throughout 5-6 day culture period [39] |
| EDC/NHS Chemistry | Cross-linking reagents for amine coupling | SPR immobilization | Activates carboxyl groups for covalent ligand attachment [41] |
In the field of antimicrobial resistance research, the dichotomy between phenotypic and genotypic testing frameworks presents a critical pathway for diagnostic and therapeutic development. While phenotypic testing measures observable microbial responses to agents, genotypic methods detect specific genetic markers associated with resistance mechanisms, offering the potential for earlier intervention and more precise characterization of resistance patterns [42]. The transition from traditional Sanger sequencing to advanced next-generation sequencing (NGS) and microarray technologies represents a paradigm shift in how researchers approach intrinsic resistance profiling, enabling higher throughput analysis and more comprehensive genetic variant detection [42]. This application note details the methodologies, protocols, and comparative analysis of three cornerstone genotyping platforms—Sanger sequencing, PCR-based approaches, and microarray systems—within the context of phenotypic versus genotypic intrinsic resistance testing research. By providing detailed experimental protocols and analytical frameworks, this document serves as a practical resource for researchers and drug development professionals seeking to implement or optimize genotyping workflows in resistance studies.
Comparative Analysis of Genotyping Platforms
Table 1: Technical and Performance Comparison of Major Genotyping Platforms
| Parameter | Sanger Sequencing | PCR-Based Methods | Microarray Genotyping |
|---|---|---|---|
| Fundamental Principle | Dideoxy chain termination sequencing [43] | Amplification of target DNA sequences using specific primers [44] | Hybridization of DNA to probe arrays fixed on a solid surface [45] |
| Throughput Capability | Low to medium (~1,000 reactions/day) [43] | Medium to high (varies by format) [43] [44] | Very high (500,000–2 million SNPs per array) [45] |
| Mutation Detection Scope | Known and unknown SNPs; determines exact base change and location [43] | Known SNPs only (e.g., TaqMan, KASP) [43] [46] | Known SNPs only (pre-designed panels) [45] |
| Accuracy | Highest ("gold standard"), near 100% detection rate [43] | High (e.g., >95% for LDR, SNaPshot, MassArray) [43] | High, but susceptible to ascertainment bias [45] [43] |
| Typical Cost Profile | High cost for large-scale studies [43] | Cost-effective for low to medium plexity [46] | Economical for high-density genotyping [45] |
| Best Suited Applications | Small-scale projects with high accuracy needs; validating NGS/microarray findings [43] | Focused studies on known markers; marker-assisted selection [43] [46] | Genome-wide association studies (GWAS); large-scale population screening [45] [47] |
| Key Limitations | Low throughput, high cost per sample for large studies [43] | Limited multiplexing without specialized designs; not for discovery [43] [44] | Cannot identify variants not pre-loaded on the array (ascertainment bias) [45] [43] |
Detailed Methodologies and Protocols
Sanger Sequencing for Resistance Marker Identification
Sanger sequencing remains the gold standard for accuracy in genetic analysis, providing definitive determination of nucleotide sequences and enabling discovery of previously unknown single nucleotide polymorphisms (SNPs) [43]. This methodology is particularly valuable in resistance research for characterizing novel resistance mutations and validating variants detected through other high-throughput methods.
Experimental Protocol for Sanger Sequencing:
- PCR Amplification: Design sequence-specific primers flanking the genomic region of interest (e.g., a segment of a viral or bacterial gene known to harbor resistance mutations). Perform PCR amplification using a thermostable DNA polymerase, dNTPs, and the target DNA template.
- Purification: Clean the amplified PCR products to remove excess primers, dNTPs, and enzymes. This can be achieved using enzymatic ExoSAP treatment or solid-phase reversible immobilization (SPRI) bead-based purification.
- Sequencing Reaction: Set up the Sanger sequencing reaction containing:
- Purified PCR product (template)
- Sequencing primer
- DNA polymerase
- Buffer
- Fluorescently labeled dideoxynucleotides (ddNTPs) and deoxynucleotides (dNTPs)
- Thermal Cycling: Run the reaction in a thermal cycler with a program designed for linear amplification, typically involving 25-35 cycles of denaturation, primer annealing, and extension.
- Purification: Remove unincorporated dye terminators from the reaction products, often using ethanol/EDTA precipitation or column-based purification.
- Capillary Electrophoresis: Load the purified products onto a capillary electrophoresis instrument. The fragments are separated by size, and the fluorescent label on each terminating ddNTP is detected by a laser, generating a chromatogram.
- Data Analysis: Use specialized software (e.g., Sequencher, Geneious) to base-call the sequence from the chromatogram and compare it to a reference sequence to identify variants.
PCR-Based Genotyping Techniques
PCR-based genotyping encompasses a range of techniques from simple allele-specific PCR to more complex digital PCR (dPCR), all leveraging the specificity of primer binding to discriminate between genetic variants [48] [49]. These methods are ideal for high-throughput screening of a limited number of predefined mutations, such as tracking known resistance markers in pathogen populations or conducting marker-assisted selection in breeding programs [46].
Experimental Protocol for Digital PCR (dPCR) for Rare Mutation Detection:
dPCR is a third-generation PCR technology that provides absolute quantification of nucleic acid targets without the need for a standard curve, offering superior sensitivity for detecting rare resistance mutations in a mixed sample [49].
- Sample Preparation: Isolate and quantify DNA from the sample (e.g., patient plasma, bacterial culture).
- Reaction Mix Preparation: Prepare a master mix containing the DNA template, fluorescently labeled target-specific probes (e.g., TaqMan probes), primers, dNTPs, and a DNA polymerase in a reaction buffer.
- Partitioning: Divide the reaction mixture into thousands to millions of discrete partitions (nanolitre-sized water-in-oil droplets or microchambers on a chip). This step is stochastic, resulting in partitions containing zero, one, or a few target molecules.
- Endpoint Amplification: Perform PCR amplification on the partitioned sample. In partitions containing the target sequence, amplification occurs, generating a fluorescent signal.
- Fluorescence Reading: After thermocycling, analyze each partition for fluorescence using a dedicated reader. Partitions are scored as positive or negative for the target signal.
- Absolute Quantification: Apply Poisson statistics to the ratio of positive to negative partitions to calculate the absolute concentration of the target sequence in the original sample, enabling precise detection of low-frequency variants [49].
Table 2: Key Research Reagent Solutions for PCR-Based Genotyping
| Reagent/Kit | Function in Protocol | Example Application in Resistance Research |
|---|---|---|
| TaqMan SNP Genotyping Assays [44] | Fluorescent probe-based detection of specific alleles during PCR. | Screening for known single nucleotide polymorphisms (SNPs) conferring drug resistance in pathogens. |
| KASP Assay Mix [46] | Kompetitive Allele-Specific PCR for bi-allelic scoring of SNPs. | Genotyping fungal strains for Fusarium wilt resistance alleles in plant breeding for marker-assisted selection. |
| dPCR Reagent Kits [49] | Optimized reagents for droplet or chip-based digital PCR. | Absolute quantification and detection of rare drug-resistant viral variants (e.g., HIV-1) in a patient sample background. |
| High-Fidelity DNA Polymerase | Accurate DNA amplification with low error rates. | Amplification of target genomic regions prior to sequencing or other downstream genotyping applications. |
Microarray-Based Genotyping
Microarray genotyping, or SNP-array technology, enables the simultaneous interrogation of hundreds of thousands to millions of genetic variants across the genome [45] [47]. The technology is based on the hybridization of fluorescently labeled DNA fragments to complementary probes immobilized on a solid surface.
Experimental Protocol for SNP Microarray Genotyping:
- DNA Extraction and Quality Control: Isolate genomic DNA and assess its quantity and quality using spectrophotometry or fluorometry.
- Fragmentation and Labeling: Fragment the DNA enzymatically or by sonication to a consistent size range. Then, label the fragmented DNA with a fluorescent dye.
- Hybridization: Mix the labeled DNA with a hybridization buffer and apply it to the microarray chip. The chip is incubated under stringent conditions to allow the target DNA to bind specifically to its complementary probes.
- Washing: Remove non-specifically bound DNA through a series of washes, reducing background signal.
- Scanning: Image the microarray using a high-resolution laser scanner that excites the fluorescent dyes and measures the intensity of emitted light at each probe location.
- Data Analysis: Use specialized software to convert fluorescence intensity data into genotype calls (AA, AB, BB) for each SNP. This data can then be used for association studies, population genetics, or profiling resistance genes across many samples simultaneously [45] [47].
Table 3: Key Research Reagent Solutions for Microarray Genotyping
| Reagent/Kit | Function in Protocol | Example Application in Resistance Research |
|---|---|---|
| Axiom Precision Medicine Array [44] | Pre-designed chip with content relevant to disease research and pharmacogenomics. | Profiling host genetic factors influencing response to antimicrobial therapy. |
| DNA Labeling Kit | Incorporates fluorescent nucleotides into fragmented DNA. | Preparing sample for hybridization in microarray workflow. |
| Hybridization Buffer & Controls | Creates optimal chemical environment for specific probe binding; controls monitor assay performance. | Ensuring specific and efficient hybridization of sample DNA to array probes. |
| Microarray Scanner | Detects fluorescence signal intensity from each probe on the array. | Generating raw data images for genotype calling. |
The selection of an appropriate genotyping methodology is paramount in designing robust phenotypic versus genotypic intrinsic resistance studies. Sanger sequencing offers unparalleled accuracy for confirmation and small-scale analysis, PCR-based methods provide cost-effective and sensitive solutions for targeted screening, and microarray platforms deliver comprehensive, high-throughput genotyping for discovery and large-scale profiling. The ongoing evolution of these technologies, particularly the rising prominence of digital PCR and next-generation sequencing, continues to enhance the resolution and scale at which resistance mechanisms can be characterized. By aligning the technical capabilities of each platform with specific research objectives—whether validating a novel mutation, monitoring known markers in a population, or conducting unbiased genome-wide scans—researchers can effectively bridge the gap between genotype and phenotype, accelerating the development of novel therapeutic strategies and diagnostic tools in the fight against antimicrobial resistance.
Next-generation sequencing (NGS) has revolutionized the field of microbial genomics, providing unprecedented capabilities for analyzing genetic determinants of antibiotic resistance. In the context of phenotypic versus genotypic intrinsic resistance testing research, NGS technologies enable comprehensive detection of resistance markers, from single nucleotide polymorphisms to complex structural variations [50]. The advent of both short-read and long-read sequencing platforms offers complementary approaches for deciphering the complete genetic basis of resistance mechanisms. Short-read technologies provide high accuracy for base-level changes, while long-read technologies excel at resolving repetitive regions and structural variations that are often implicated in resistance [26] [51]. This application note details experimental protocols and analytical frameworks for implementing these advanced genomic tools in antimicrobial resistance research.
Next-generation sequencing technologies have evolved into two principal categories, each with distinct strengths and limitations critical for resistance research.
Short-read sequencing (e.g., Illumina, MGI DNBSEQ) is characterized by parallel sequencing of millions of small DNA fragments (typically 50-600 base pairs) [52] [50]. These platforms achieve exceptional base-level accuracy (>99.9%) and high throughput at a low cost per base, making them ideal for detecting single nucleotide variants (SNVs) and small insertions/deletions (indels) associated with antibiotic resistance [50] [51]. However, their limited read length poses challenges for assembling repetitive genomic elements and resolving complex structural variations.
Long-read sequencing (e.g., PacBio Single Molecule Real-Time [SMRT] sequencing, Oxford Nanopore Technologies [ONT]) generates reads that can span thousands to tens of thousands of bases [52] [50]. This capability allows for direct sequencing through repetitive regions, transposable elements, and resistance gene clusters, providing a complete picture of genomic architecture [51]. While historically burdened by higher error rates (5-15%), recent advancements such as PacBio HiFi circular consensus sequencing and ONT's improved base-calling have significantly enhanced accuracy, making these technologies increasingly viable for detecting resistance variants [26] [51].
Table 1: Comparative Analysis of Short-Read and Long-Read Sequencing Technologies
| Feature | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Representative Platforms | Illumina (ISeq100, MiSeq), MGI DNBSEQ-G400 [50] [26] | PacBio SMRT, Oxford Nanopore (MinION, GridION) [50] [26] |
| Typical Read Length | 50-600 base pairs [52] [50] | 10,000-30,000+ base pairs [50] |
| Primary Strengths | High base-level accuracy, low cost per base, high throughput [50] [51] | Resolution of structural variants, haplotype phasing, direct epigenetic detection [50] [51] |
| Error Mode | Mainly substitution errors [50] | Mainly insertion-deletion errors [50] |
| Ideal for Resistance Research | SNV calling, small indels, comprehensive variant detection in coding regions [26] | Complex structural variations, repetitive resistance gene contexts, full-length plasmid sequencing [26] [51] |
Integrated Experimental Protocols
Sample Preparation and Nucleic Acid Extraction
The initial step for all NGS workflows is the isolation of high-quality genetic material. The required yield, purity, and integrity of nucleic acids are critical for successful library construction [53].
- Sample Types: Microbial cultures, clinical isolates (e.g., sputum for Mycobacterium tuberculosis), or purified plasmids can serve as source material [54] [26].
- Extraction Protocol: Use commercial pathogen-specific kits (e.g., Roche MagNA Pure) for DNA/RNA extraction. For RNA viruses, perform reverse transcription to generate cDNA [26].
- Quality Control (QC): Assess DNA/RNA integrity via agarose gel electrophoresis or microfluidic analysis (e.g., TapeStation). Quantify nucleic acids using fluorometric methods (e.g., Qubit Flex) to ensure accurate measurements. Verify purity via UV spectrophotometry (A260/A280 ratio ~1.8-2.0) [53].
Library Preparation for Short-Read Sequencing
Library construction converts fragmented nucleic acids into a sequenceable format [53]. The following protocol is adapted for bacterial whole-genome sequencing for resistance studies.
Table 2: Key Research Reagent Solutions for Short-Read Library Preparation
| Reagent | Function | Example Product |
|---|---|---|
| Fragmentation Enzyme | Enzymatically shears DNA to desired size (e.g., 300-500 bp). | DeepChek NGS Library Prep Kit [26] |
| Adapter Oligos | Ligate to fragment ends; contain sequences for flow cell binding and indexing. | Illumina P5/P7 Adapters [53] |
| High-Fidelity DNA Polymerase | Amplifies the adapter-ligated library with minimal bias. | DeepChek Library Amplification Mix [26] |
| Size Selection Beads | Purify libraries and select for fragments of the desired size range. | AMPure XP Beads [26] |
Workflow Steps:
- Fragmentation: Use 10-100 ng of genomic DNA. Fragment via enzymatic treatment (37°C for 30 minutes) to a target size of 300-500 bp [26].
- End Repair and A-Tailing: Repair fragment ends and add an 'A' base overhang to facilitate adapter ligation (20°C for 30 min, then 65°C for 30 min) [26].
- Adapter Ligation: Ligate indexed adapter sequences to the fragments (20°C for 15 min). Adapters enable binding to the sequencer flow cell and allow for sample multiplexing [53] [26].
- Library Amplification: Perform 8 cycles of PCR to enrich for properly ligated fragments using a high-fidelity polymerase [26].
- Purification and QC: Clean the final library using size-selection beads (e.g., 0.8x ratio of AMPure XP beads). Validate library quality and concentration using a fragment analyzer (e.g., TapeStation) and fluorometry [26].
Library Preparation for Long-Read Sequencing
Long-read library preparation focuses on preserving and sequencing high-molecular-weight DNA.
Workflow Steps:
- DNA Quality: Use high-integrity genomic DNA. Avoid methods that cause excessive shearing.
- Size Selection (Optional): For targeting very long reads, size-select DNA fragments >20 kb using pulsed-field gel electrophoresis or magnetic bead-based methods.
- Adapter Ligation: Ligate platform-specific adapters (e.g., Nanopore SQK-LSK109 ligation kit) to the DNA fragments. Transposase-based "tagmentation" methods can combine fragmentation and adapter insertion in a single step [51].
- QC: Assess the final library for fragment size distribution and concentration using a fragment analyzer.
Sequencing and Data Analysis
Sequencing Execution:
- Short-Read: Load normalized libraries onto the sequencer (e.g., Illumina MiSeq or iSeq100). Use a 2x150 bp or 2x250 bp paired-end run configuration for optimal resistance gene coverage. Include 1% PhiX control to monitor sequencing quality [26].
- Long-Read: Load the library according to the manufacturer's instructions (e.g., onto a Nanopore MinION or PacBio SMRT cell). Base-calling can be performed in real-time (Nanopore) or post-run [26].
Bioinformatic Analysis for Resistance Detection:
- Base Calling and Demultiplexing: Convert raw signals to nucleotide sequences (FASTQ files) and separate data by sample using barcode information [53].
- Quality Control and Trimming: Use tools like FastQC to assess read quality. Trim low-quality bases and adapter sequences with Trimmomatic or Cutadapt.
- Alignment/Assembly: For short-read data, align reads to a reference genome using BWA or Bowtie2. For long-read data, perform de novo genome assembly using Canu or Flye to reconstruct complete genomes and plasmids [26].
- Variant Calling: Identify single nucleotide polymorphisms (SNPs) and indels relative to a reference using tools like DeepVariant. For hybrid sequencing data, specialized models that combine short- and long-read inputs can improve variant detection accuracy, especially in challenging genomic regions [55].
- Resistance Gene Identification: Use assembled genomes or aligned reads as input for specialized resistance databases and tools (e.g., CARD, Mykrobe, DeepChek) to identify known resistance mutations and acquired resistance genes [26].
Application in Resistance Research: A Case Study
A recent study demonstrated the application of both short-read and long-read NGS for comprehensive detection of drug resistance mutations across multiple pathogens, including HIV, HBV, HCV, and Mycobacterium tuberculosis [26].
Experimental Design:
- Samples: 15 quality control samples from various pathogens.
- Platforms: Short-read (Illumina ISeq100, MiSeq; MGI DNBSEQ-G400) and long-read (Oxford Nanopore MinION).
- Method: Target-specific amplification of resistance-associated genomic regions (e.g., HIV protease/reverse transcriptase) followed by sequencing on all platforms.
- Analysis: Unified bioinformatic processing using DeepChek software for variant calling.
Key Findings:
- High Concordance: The study demonstrated high concordance for majority mutations across all four NGS platforms.
- Minority Variant Detection: Nanopore technology detected a higher number of low-frequency minority variants (<20% frequency), which are crucial for understanding evolving resistance in quasispecies populations like HIV [26].
- Utility of Hybrid Approaches: Combining data from different technologies or using hybrid analysis models can improve detection accuracy. For instance, a hybrid DeepVariant model that jointly processes Illumina and Nanopore data has been shown to improve germline variant detection, a strategy that can be adapted for detecting resistance variants in bacterial populations [55].
Short-read and long-read NGS technologies provide powerful, complementary tools for advancing research into phenotypic and genotypic intrinsic resistance. While short-read platforms remain the workhorse for high-accuracy detection of single-nucleotide resistance mutations, long-read technologies are unparalleled for resolving the complex genomic architectures that underlie many resistance mechanisms. The protocols and case studies outlined herein provide a framework for researchers to implement these advanced genomic tools. A combined, hybrid approach leveraging both technologies, supported by robust bioinformatic analysis, offers the most comprehensive strategy for elucidating the full genetic basis of antimicrobial resistance.
Antimicrobial resistance (AMR) presents a critical global health threat, projected to claim over 39 million lives worldwide in the next 25 years [56]. The cornerstone of effective antimicrobial stewardship is timely and accurate antimicrobial susceptibility testing (AST), which has traditionally operated within a paradigm tension between genotypic and phenotypic methods. Phenotypic resistance describes the observable resistance of a bacterial population to an antibiotic, typically measured through minimum inhibitory concentration (MIC) assays that determine the lowest antibiotic concentration required to inhibit bacterial growth [5]. In contrast, genotypic resistance refers to the presence of specific genetic determinants that confer resistance potential, such as resistance genes (mecA, vanA/B, blaKPC) or mutations [5] [32].
While molecular genotypic methods offer rapid turnaround times (1-5 hours), they face significant limitations: they detect only pre-defined resistance mechanisms and may miss novel or complex resistance patterns [14] [32]. For Gram-negative bacteria particularly, resistance mechanisms are heterogeneous, and the absence of a detected AMR gene does not always equate to phenotypic susceptibility [32]. Phenotypic testing remains the clinical gold standard as it directly measures the functional interaction between bacteria and antimicrobials [5]. Next-generation rapid phenotypic AST (RAST) technologies bridge this divide by offering the comprehensive assessment of phenotypic methods with significantly reduced turnaround times, potentially revolutionizing clinical microbiology practice and patient outcomes [14] [57].
Commercial Rapid Phenotypic AST Platforms
The rapid AST landscape has expanded considerably, with multiple platforms receiving regulatory approval. These systems employ diverse technological approaches to accelerate phenotypic susceptibility testing directly from positive blood cultures, substantially reducing time-to-result compared to conventional methods that require 18-24 hours of incubation [57].
Table 1: Commercial Rapid Phenotypic AST Platforms
| Platform (Manufacturer) | Technology Principle | Time-to-Result | Regulatory Status (as of 2025) | Key Applications |
|---|---|---|---|---|
| Accelerate Pheno system (Accelerate Diagnostics) | Morphokinetic Cellular Analysis (MCA) using time-lapse dark field microscopy and machine learning [57] | ~7 hours from sample loading [57] | FDA-cleared (since 2017), CE-marked [57] | Combined identification and AST directly from positive blood cultures [57] |
| VITEK REVEAL (bioMérieux) | Detection of volatile organic compounds (VOCs) released during bacterial metabolism [58] | ~6.5 hours mean [59] [58] | FDA Breakthrough Device, 510(k) cleared in 2024 [56] | AST directly from positive blood cultures for Gram-negative bacteria [59] |
| ASTar (Q-linea) | High-speed time-lapse microscopy imaging of bacteria [56] | Not specified in results | FDA 510(k) cleared in 2024 [56] | AST from positive blood cultures [57] |
| LifeScale (Affinity Biosensors) | Microfluidics and AI to measure bacterial replication and MICs [56] | Not specified in results | FDA 510(k) cleared in 2024 [56] | AST from positive blood cultures [56] |
Performance Characteristics of Selected Platforms
Recent head-to-head comparisons provide critical performance data for evaluating these systems. A 2025 study compared three RAST systems using 220 prospectively collected Gram-negative positive blood cultures against 25 antibiotics [59] [58].
Table 2: Performance Comparison of RAST Systems for Gram-Negative Bacteria
| Performance Metric | VITEK REVEAL | VITEK 2-RAST | DD-RAST (EUCAST) |
|---|---|---|---|
| Essential Agreement (EA) | 97.1% (3,603 combinations) [59] | 96.2% (3,941 combinations) [59] | Not applicable |
| Categorical Agreement (CA) | 98.3% [59] | 98.4% [59] | 98.2% (2,388 combinations) [59] |
| Mean Time-to-Result | 6 hours 32 minutes [59] | 13 hours 51 minutes [59] | 8 hours (fixed) [59] |
| Very Major Error (VME) Rate | ≤1.8% [59] | ≤1.8% [59] | ≤1.8% [59] |
For the Accelerate Pheno system, a multicenter evaluation demonstrated an overall categorical agreement of 97.9% for Gram-positive cocci and 94.3% for Gram-negative rods when testing positive blood cultures [57]. The system provided identification results in approximately 2 hours and AST results about 7 hours after test initiation, representing an improvement of 23-42 hours in time to results compared to standard methods [57].
Experimental Protocols
Protocol: Rapid Phenotypic AST Using the VITEK REVEAL System
Principle: The VITEK REVEAL system utilizes 96-well broth microdilution plates and detects volatile organic compounds released by metabolically active bacteria. When bacterial growth is inhibited by an effective antibiotic, VOC production decreases, allowing for rapid minimum inhibitory concentration determination [58].
Materials:
- VITEK REVEAL instrument
- BacT/Alert FA Plus (aerobic) and FN Plus (anaerobic) blood culture bottles
- BacT/Alert Virtuo automated blood culture system
- REVEAL broth microdilution plates
- MacConkey and 5% sheep blood tryptic soy agar plates
- MALDI-TOF mass spectrometry system for identification
Procedure:
- Sample Collection and Incubation: Collect blood samples into BacT/Alert culture bottles and incubate in the BacT/Alert Virtuo system until positivity [58].
- Gram Staining: Perform Gram staining on positive blood cultures to confirm monomicrobial Gram-negative infection [58].
- Species Identification: Perform direct MALDI-TOF mass spectrometry identification from positive blood culture broth [58].
- REVEAL Inoculation: Inoculate REVEAL broth microdilution plates directly from positive blood culture bottles according to manufacturer specifications [58].
- Incubation and Reading: Load plates into the VITEK REVEAL instrument for incubation and automated reading. The system monitors VOC production for 5.5-6 hours [58].
- MIC Determination: The instrument software determines MIC values based on VOC detection patterns [58].
- Result Interpretation: Compare MIC values to appropriate breakpoints (e.g., EUCAST) for categorical interpretations (Sensitive, Intermediate, Resistant) [58].
Protocol: In-Situ Time-Lapse Imaging of Microcolonies (ISM-TLI)
Principle: This method uses an automated imaging system to track microcolony growth on a 96-well gel plate containing antibiotic gradients. Image processing algorithms analyze growth rates to determine MIC within 2-3 hours [60].
Materials:
- ISM-TLI system with temperature-controlled incubation module
- Custom 96-well gel plates
- Time-lapse imaging module
- Cation-adjusted Mueller-Hinton broth
- Phosphate-buffered saline
Procedure:
- Sample Preparation: Adjust bacterial suspension to approximately 10⁶ CFU/mL in cation-adjusted Mueller-Hinton broth [60].
- Plate Inoculation: Inoculate 96-well gel plates containing antibiotic gradients with bacterial suspension [60].
- System Loading: Load plates into the ISM-TLI system [60].
- Image Acquisition: The system automatically performs time-lapse imaging of microcolony growth at 37°C [60].
- Image Analysis: Integrated algorithms perform image registration, microcolony identification, and growth rate calculation [60].
- MIC Interpretation: Determine MIC based on statistical histograms of bacterial area changes after 2-3 hours of incubation [60].
- Quality Control: Include appropriate reference strains (e.g., E. coli ATCC 25922, P. aeruginosa ATCC 27853) with each run [60].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents for Rapid Phenotypic AST
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Broth Microdilution Plates | Provide standardized antibiotic concentration gradients for MIC determination | VITEK REVEAL plates; Custom 96-well gel plates for ISM-TLI [58] [60] |
| Culture Media | Support bacterial growth during testing | Cation-adjusted Mueller-Hinton broth; Blood culture media (BacT/Alert) [58] [60] |
| Reference Strains | Quality control and method validation | ATCC strains (e.g., E. coli ATCC 25922, P. aeruginosa ATCC 27853, S. aureus ATCC 29213) [60] |
| Blood Culture Bottles | Sample collection and initial amplification | BacT/Alert FA Plus (aerobic), FN Plus (anaerobic) [58] |
| Identification Systems | Species determination essential for AST interpretation | MALDI-TOF mass spectrometry; Fluorescence in situ hybridization (FISH) [57] [58] |
Workflow and Technology Diagrams
Antimicrobial resistance (AMR) is a significant source of morbidity and mortality worldwide, with the Centers for Disease Control and Prevention (CDC) reporting that over 2 million people in the U.S. become ill with antimicrobial-resistant infections each year, resulting in more than 23,000 deaths [61]. The escalating threat of AMR, particularly from multidrug-resistant pathogens such as Acinetobacter baumannii and Pseudomonas aeruginosa, underscores the critical need for advanced diagnostic and screening methodologies in drug development [62] [63]. A comprehensive understanding of resistance mechanisms—categorized into limiting drug uptake, drug target modification, drug inactivation, and active drug efflux—is fundamental to developing novel therapeutic agents [61].
This application note details integrated experimental approaches for elucidating antimicrobial resistance mechanisms and conducting compound screening. We place specific emphasis on the comparative analysis of phenotypic and genotypic profiling of intrinsic resistance, providing detailed protocols and data analysis frameworks designed to accelerate the discovery of novel antibacterial agents.
Resistance Mechanism Elucidation: Phenotypic vs. Genotypic Profiling
Intrinsic resistance is a naturally occurring phenomenon universal within a bacterial species, independent of previous antibiotic exposure, and not related to horizontal gene transfer [64]. A core component of resistance research involves comparing traditional phenotypic methods with modern genotypic techniques for detecting resistance mechanisms, particularly in challenging pathogens.
Phenotypic Detection Methods for Metallo-β-Lactamase (MBL) Production
The following protocols are standardized for detecting MBL production in Gram-negative bacteria such as Acinetobacter baumannii.
Protocol 2.1.1: Double-Disk Synergy Test (DDST)
- Principle: This test detects MBL production by observing the enhanced zone of inhibition around a carbapenem disk in the presence of a chelating agent (EDTA) that inactivates MBLs.
- Procedure:
- Prepare a 0.5 McFarland suspension of the test isolate in saline.
- Lawn culture the suspension onto a Mueller-Hinton Agar (MHA) plate.
- Place an imipenem (10 µg) or meropenem (10 µg) disk in the center of the plate.
- Place a blank filter disk approximately 10 mm center-to-center from the antibiotic disk.
- Apply 10 µL of 0.5 M EDTA solution to the blank disk.
- Incubate the plate at 35°±2°C for 16-18 hours.
- Interpretation: A positive result is indicated by an enlarged zone of inhibition on the side of the antibiotic disk facing the EDTA disk [62].
Protocol 2.1.2: Modified Hodge Test (MHT)
- Principle: This test detects carbapenemase production by demonstrating the ability of a bacterial isolate to hydrolyze carbapenems, which allows growth of a susceptible indicator strain in a cloverleaf-like pattern.
- Procedure:
- Prepare a 1:10 dilution of a 0.5 McFarland E. coli ATCC 25922 suspension in saline.
- Lawn culture the diluted suspension onto an MHA plate.
- Place an erapenem (10 µg) or meropenem (10 µg) disk in the center of the plate.
- Streak the test isolates in a straight line from the edge of the disk to the periphery of the plate.
- Incubate at 35°±2°C for 16-18 hours.
- Interpretation: A positive result is indicated by a distorted zone of inhibition or a cloverleaf-like indentation where the test isolate streak intersects the zone of the indicator strain [62].
Protocol 2.1.3: MBL-E Test
- Principle: This test uses a predefined gradient of imipenem and EDTA on a strip to determine the Minimum Inhibitory Concentration (MIC) and confirm MBL production.
- Procedure:
- Prepare a 0.5 McFarland suspension of the test isolate.
- Lawn culture the suspension onto an MHA plate.
- Place an MBL-E strip onto the inoculated agar surface.
- Incubate at 35°±2°C for 16-18 hours.
- Interpretation: An MIC ratio of imipenem to imipenem+EDTA of ≥8 is considered positive for MBL production [62].
Genotypic Detection of Resistance Genes
Molecular methods offer a rapid and specific means for identifying resistance genes. The following protocol details the detection of key MBL genes.
Protocol 2.2.1: PCR Detection of MBL Genes
- Principle: Polymerase Chain Reaction (PCR) amplifies specific gene sequences associated with resistance, such as blaNDM, blaOXA-58, and blaVIM.
- Reagents and Equipment:
- Thermal cycler
- PCR master mix (includes Taq polymerase, dNTPs, MgCl₂)
- Primer sequences (see Table 1)
- DNA template (extracted from bacterial isolates)
- Gel electrophoresis apparatus
- Procedure:
- DNA Extraction: Extract genomic DNA from pure bacterial cultures using a commercial bacterial DNA extraction kit.
- PCR Reaction Setup:
- Prepare a 25 µL reaction mixture containing:
- 12.5 µL of PCR master mix
- 1 µL each of forward and reverse primer (10 µM concentration)
- 2 µL of DNA template
- 8.5 µL of nuclease-free water
- Prepare a 25 µL reaction mixture containing:
- Amplification Conditions:
- Initial Denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 55-60°C (primer-specific) for 30 seconds
- Extension: 72°C for 1 minute per kb
- Final Extension: 72°C for 7 minutes
- Amplification Product Analysis:
- Subject PCR products to gel electrophoresis (1.5% agarose).
- Visualize under UV transillumination after staining with ethidium bromide.
- Interpretation: The presence of a band of the expected size (as defined by the primer set) confirms the presence of the target resistance gene [62].
Comparative Data Analysis: Phenotypic vs. Genotypic Methods
A recent prospective cross-sectional study on Acinetobacter baumannii provides quantitative data comparing the efficacy of these methodological approaches, summarized in the table below [62].
Table 1: Comparison of MBL Detection Methods in A. baumannii (n=93 drug-resistant isolates)
| Detection Method | Type | Positive Isolates (n) | Detection Rate (%) | Key Findings/Limitations |
|---|---|---|---|---|
| Double-Disk Synergy Test | Phenotypic | 34 | 36.54% | Lower sensitivity but high specificity. |
| Modified Hodge Test | Phenotypic | 83 | 89.42% | High sensitivity but may yield false positives. |
| MBL-E Test | Phenotypic | 69 | 74.19% | Provides quantitative MIC data. |
| PCR (NDM, OXA-58, VIM) | Genotypic | 56 | 60.00% | Gold standard for specific gene detection; faster but may miss novel genes. |
Table 2: Primer Sequences for PCR Detection of Key MBL Genes
| Target Gene | Primer Sequence (5' to 3') | Amplicon Size (bp) | Reference |
|---|---|---|---|
| blaNDM | F: AACACAGCCTGACTTTCGR: TGATATTGTCACTGGTGTGG | ~300 | [62] |
| blaOXA-58 | F: TGGCACGCATTTAGACCGR: AAACCCACATACCAACCC | ~200 | [62] |
| blaVIM | F: GATGGTGTTTGGTCGCATAR: CGAATGCGCAGCACCAG | ~500 | [62] |
Advanced Compound Screening Workflows
Overcoming intrinsic resistance requires the discovery of novel compounds with activity against multidrug-resistant pathogens. Modern screening leverages automation, artificial intelligence (AI), and complex physiological models.
High-Throughput Screening (HTS) of Small Molecules
Protocol 3.1.1: Automated Primary Screening Assay
- Objective: To identify "hit" compounds with inhibitory activity against a molecular target or bacterial growth from large compound libraries.
- Reagents and Equipment:
- Beckman Coulter Biomek i-Series Automated Liquid Handler
- Beckman Coulter Echo Acoustic Liquid Handler
- Compound library (e.g., 10^6 compounds)
- Assay plates (96, 384, or 1536-well)
- IDBS Polar Biopharma Lifecycle Management Software
- Target protein or bacterial strain
- Detection reagents (e.g., fluorescence-based)
- Procedure:
- Compound Management: Use the Echo Acoustic Liquid Handler to dispense nanoliter volumes of compounds from stock solutions into assay plates. Track compound integrity and location using IDBS Polar software [65].
- Assay Plate Preparation: Using the Biomek i-Series Automated Liquid Handler, add the target (e.g., purified enzyme, bacterial suspension) and reagents to the compound plates in a buffer suitable for the reaction [65] [66].
- Incubation and Reaction: Incubate the plates under optimal conditions (e.g., 37°C for 1-2 hours) to allow the reaction.
- Signal Detection: Measure the assay endpoint using a plate reader, imager, or flow cytometer. Readouts can include fluorescence, luminescence, or absorbance [65] [66].
- Data Analysis: Use specialized software like GeneData Screener for statistical analysis of large datasets. Calculate % inhibition relative to positive (no compound) and negative (no activity) controls. Compounds showing significant activity (typically >50-70% inhibition) are designated as "hits" [65].
AI-Driven de Novo Antibiotic Design
A groundbreaking generative AI framework demonstrates a powerful alternative to traditional library screening by designing novel antibiotic molecules from scratch [67].
Protocol 3.2.1: Generative AI for Antibiotic Design
- Principle: Two AI models are used to explore vast chemical spaces beyond existing compound libraries:
- Fragment-based (CReM): Starts with active molecular fragments and computationally expands them.
- Unconstrained de novo (VAE): Directly generates entirely new molecular structures without a starting fragment.
- Workflow:
- Model Training: Train the AI models on datasets of molecules with known antibacterial activity and cytotoxicity.
- Compound Generation: Generate millions of candidate molecules with predicted antibacterial activity.
- In Silico Filtering: Filter candidates for desirable drug-like properties.
- Synthesis and Validation: Select top candidates for chemical synthesis and experimental validation in phenotypic assays [67].
- Outcome: This approach led to the design of compounds NG1 and DN1, which demonstrated efficacy against multidrug-resistant Neisseria gonorrhoeae and Staphylococcus aureus in mouse models, with a unique mechanism of action and rapid bactericidal activity [67].
Phenotypic Screening Using 3D Organoid Models
For evaluating compound efficacy and toxicity in a physiologically relevant environment, 3D organoid models are increasingly employed.
Protocol 3.3.1: Tumoroid Screening for Anti-Cancer Compounds
- Principle: Patient-derived tumor organoids (tumoroids) mimic the original tumor's morphology, genetics, and drug response, enabling more predictive in vitro screening.
- Reagents and Equipment:
- Molecular Devices CellXpress.AI Automated Cell Culture System
- Confocal microscope (e.g., Leica Microsystems Stellaris)
- FLIPR Penta System for calcium flux assays
- Primary tumor cells or stem cells
- Procedure:
- Tumoroid Generation: Culture patient-derived tumor cells in a 3D extracellular matrix with specific growth factors to promote self-organization into tumoroids. This process can take weeks [68].
- Automated Maintenance and Screening: Use the automated CellXpress.AI system to handle media exchanges, compound addition, and monitoring. This ensures consistency and scalability [68].
- Compound Treatment: Treat tumoroids with candidate compounds in a 96-well format.
- Multiparametric Analysis:
- Viability Assay: Measure cell viability using ATP-based assays.
- High-Content Imaging: Use confocal microscopy to obtain 3D images of tumoroids. Analyze structure, cell death, and proliferation markers.
- Functional Assays: Use dyes and the FLIPR Penta System to measure functional responses like calcium oscillations [68].
- Data Analysis: Employ machine learning algorithms to analyze complex, multiparametric data and distinguish compound-induced phenotypes [68].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Resistance and Screening Studies
| Item | Function/Application | Example |
|---|---|---|
| Mueller-Hinton Agar | Standardized medium for antimicrobial susceptibility testing (AST). | [62] |
| MBL-E Test Strips | Phenotypic confirmation and MIC determination for Metallo-β-Lactamases. | [62] |
| PCR Master Mix & Primers | Genotypic detection of specific resistance genes (e.g., blaNDM, blaVIM). | [62] |
| Automated Liquid Handlers | Precise, high-throughput dispensing of compounds and reagents in screening assays. | Beckman Coulter Echo & Biomek i-Series [65] |
| IDBS Polar Software | Biopharma lifecycle management for tracking compounds and assay data. | [65] |
| GeneData Screener | Statistical analysis and management of high-throughput screening data. | [65] |
| Confocal Microscope | High-resolution imaging for complex assays, including 3D organoid analysis. | Leica Microsystems Stellaris [68] |
| FLIPR Penta System | High-throughput cellular screening for functional assays (e.g., calcium flux). | Molecular Devices [68] |
Workflow Visualizations
Resistance Mechanism Elucidation Workflow
Compound Screening and Validation Workflow
The integrated application of phenotypic and genotypic profiling provides a powerful strategy for deciphering the complex landscape of antimicrobial resistance. As demonstrated, phenotypic methods like the MBL-E test offer functional insights, while genotypic PCR provides rapid, specific gene detection [62]. The synergy of these approaches is critical for understanding intrinsic resistance mechanisms, such as outer membrane impermeability and efflux pump activity, which are major contributors to the multidrug-resistant phenotype in Gram-negative pathogens [64].
Complementing this, modern compound screening—spanning from automated HTS and generative AI design to physiologically relevant 3D organoid models—offers a multifaceted pipeline for discovering and validating novel therapeutic agents against resistant infections [65] [67] [68]. The adoption of these detailed and standardized protocols, supported by advanced reagent solutions and data analysis tools, provides a robust framework for researchers to accelerate the development of effective treatments against the escalating threat of antimicrobial resistance.
Navigating Challenges: Technical Limitations and Implementation Strategies
Genotype-phenotype discordance in antimicrobial resistance (AMR) profiling presents a significant challenge in clinical microbiology and pharmaceutical development. This phenomenon occurs when detectable genetic resistance markers fail to correlate with observable phenotypic resistance patterns, potentially leading to inappropriate treatment decisions. While rapid molecular diagnostics provide accelerated pathogen identification and resistance detection, discrepancies with conventional phenotypic methods remain a critical concern. This application note examines the sources of these discrepancies across bacterial and viral pathogens, provides systematic protocols for their investigation, and highlights emerging computational approaches that enhance prediction accuracy. By integrating validated laboratory methods with advanced analytical frameworks, researchers can better navigate the complexities of resistance prediction, ultimately supporting more effective therapeutic development and clinical management.
The escalating crisis of antimicrobial resistance has intensified reliance on both genotypic and phenotypic testing methods to guide therapeutic decisions. Traditional culture-based phenotypic methods, while considered the gold standard, require 48-72 hours to yield results, potentially delaying optimal treatment [32]. Rapid molecular diagnostics reduce this turnaround time to hours by detecting specific genetic resistance markers, enabling earlier targeted therapy [32]. However, an important limitation emerges when discordance arises between these rapid genotypic results and subsequent phenotypic antimicrobial susceptibility testing (AST) of recovered isolates [32].
This genotype-phenotype discordance represents a critical challenge across clinical and research settings. For bacterial pathogens, discordance may stem from undetected resistance mechanisms, complex genetic interactions, or expression variability [69]. In viral pathogens like HIV, complex mutation patterns can yield discrepant interpretations between genotypic and phenotypic resistance testing [70]. Understanding and addressing these discrepancies is essential for accurate resistance prediction, effective therapeutic development, and improved patient outcomes.
Quantitative Analysis of Discordance Patterns
Bacterial Resistance Discordance in Escherichia coli
A comprehensive comparison of whole-genome sequencing (WGS) and broth microdilution methods for 234 E. coli bloodstream isolates revealed substantial variation in categorical agreement across antibiotic classes [69]. The study evaluated concordance between genotypic resistance markers and phenotypic susceptibility results using standardized breakpoints.
Table 1: Categorical Agreement Between Genotypic and Phenotypic AST for E. coli
| Antibiotic | Categorical Agreement | Discordance Pattern |
|---|---|---|
| Gentamicin | 100% | Perfect agreement |
| Meropenem | 100% | No resistance observed |
| Amikacin | >95% | High agreement |
| Tobramycin | >95% | High agreement |
| Cefepime | >95% | High agreement |
| Cefotaxime | >95% | High agreement |
| Ceftazidime | >95% | High agreement |
| Amoxicillin | >95% | High agreement |
| Amoxicillin/Clavulanic Acid | <95% | Notable discordance |
| Piperacillin/Tazobactam | <95% | Notable discordance |
| Ciprofloxacin | <95% | Notable discordance |
Most discrepancies occurred in isolates with minimum inhibitory concentrations (MICs) within ±1 doubling dilution of clinical breakpoints, highlighting the impact of technical variability in borderline cases [69]. Additionally, 22.73% of major errors (resistant genotype/susceptible phenotype) represented isolates that tested phenotypically susceptible at higher antibiotic exposures, categorized as "not resistant" rather than fully susceptible [69].
Methodological Comparisons for Resistance Prediction
Different methodological approaches demonstrate varying capabilities for predicting resistance phenotypes from genetic information.
Table 2: Comparison of AMR Prediction Methodologies
| Methodology | Target | Accuracy Metrics | Limitations |
|---|---|---|---|
| Whole Genome Sequencing (WGS) | Acquired resistance genes, chromosomal mutations | 95-100% agreement for specific drug-bug combinations [69] | Limited by current knowledge of resistance mechanisms; lower accuracy for ciprofloxacin, beta-lactam/inhibitor combinations |
| 16S rRNA Functional Prediction (PICRUSt2, Tax4Fun) | Imputed gene content from taxonomic profiles | F1 scores: 0.08-0.22 for carbapenem resistance in E. coli [71] | Low accuracy for clinical AMR prediction; fails to detect horizontally acquired resistance |
| Transcriptomic ML Classifiers | Gene expression signatures | 96-99% accuracy for P. aeruginosa with 35-40 gene sets [72] | Requires antibiotic-specific training; limited to known resistance phenotypes |
| HIV Genotypic Testing | Reverse transcriptase and protease mutations | 80-90% concordance with phenotypic testing [70] | Difficult to interpret complex mutation patterns; 10-20% require phenotypic clarification |
Experimental Protocols for Discordance Investigation
Protocol 1: Resolution of Bacterial AST Discordance
Principle: Systematically investigate discrepancies between detected resistance genes and phenotypic AST results to identify novel mechanisms or technical artifacts [32].
Materials:
- Bacterial isolate with demonstrated genotype-phenotype discordance
- Appropriate agar and broth media for the organism
- Antibiotic powders for gradient concentration preparation
- PCR reagents for amplification of suspected resistance genes
- DNA sequencing capabilities for mutation detection
- Equipment for standard AST (broth microdilution, disk diffusion, or automated systems)
Procedure:
- Confirm Phenotypic Results: Repeat AST using reference broth microdilution method according to EUCAST or CLSI standards [69]. Include quality control strains.
- Verify Genotypic Detection: Repeat PCR or re-extract DNA and sequence relevant genetic regions. For WGS data, verify bioinformatics pipeline parameters and database versions.
- Investigate Mixed Cultures: Subculture to isolate individual colonies, as polymicrobial infections may associate resistance genes with different organisms [32].
- Screen for Additional Mechanisms: For β-lactam discordance, perform double-disk synergy tests for extended-spectrum β-lactamases (ESBLs) or modified carbapenem inactivation method (mCIM) for carbapenemases.
- Evaluate Gene Expression: For genes detected without phenotypic correlation, perform RT-PCR to assess expression levels under standardized conditions.
- Examine Genetic Context: For plasmid-mediated resistance, attempt plasmid transfer via conjugation to assess mobility and expression in new hosts.
Interpretation: Persistent phenotypic resistance without genetic explanation suggests novel mechanisms. Detected genes without phenotypic correlation may indicate silent resistance determinants or technical detection artifacts.
Protocol 2: HIV-1 Phenotypic Drug Susceptibility Testing
Principle: Measure direct antiviral effects of inhibitors on patient-derived HIV-1 strains using recombinant lentivirus technology with dual-reporter systems [70].
Materials:
- Patient HIV-1 RNA or DNA containing pol gene
- Dual-reporter lentivirus transfer plasmid (luciferase and ZsGreen)
- Lentivirus packaging system (envelope and packaging plasmids)
- 293FT and 293A cell lines
- Antiretroviral drugs in purified powder form
- Luciferase assay system
- Fluorescence microscopy for ZsGreen detection
Procedure:
- Amplify Patient pol Gene: Extract viral RNA/DNA and amplify full-length protease and reverse transcriptase genes using RT-PCR/PCR with appropriate primers.
- Clone into Packaging System: Insert patient pol gene into modified lentivirus packaging plasmid (psPAX2m-Pol) [70].
- Generate Recombinant Lentivirus: Co-transfect 293FT cells with packaging, envelope, and transfer plasmids using standard methods.
- Titer Virus Stocks: Determine viral titer by transducing 293A cells and measuring ZsGreen-positive cells via fluorescence microscopy or flow cytometry.
- Optimize Assay Conditions: Establish optimal multiplicity of infection (MOI = 10), cell density (1.5×10^4 cells/well), and incubation time (60 hours) using wild-type control virus [70].
- Perform Drug Susceptibility Testing: Incurate diluted drugs with recombinant virus and 293A cells across concentration ranges. Include wild-type virus as control.
- Quantify Results: Measure luciferase activity and calculate IC50 values. Determine fold-change (FC) relative to wild-type virus.
Interpretation: FC < 1.5 = susceptible; FC 1.5-3.5 = low-level resistance; FC 3.5-10 = intermediate resistance; FC > 10 = high-level resistance [70].
Visualization of Discordance Resolution Pathways
Gram-Negative Discordance Investigation
Diagram 1: Gram-Negative Discordance Workflow
Machine Learning Resistance Prediction
Diagram 2: ML-Based Resistance Prediction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Discordance Investigation
| Reagent/System | Application | Function | Example Use |
|---|---|---|---|
| Broth Microdilution Panels | Phenotypic AST | Gold standard quantification of MIC | EUCAST-standardized panels for Enterobacterales [69] |
| Dual-Reporter Lentivirus System | Viral phenotyping | Measures drug susceptibility with luciferase/ZsGreen | HIV-1 pol gene susceptibility testing [70] |
| WGS Bioinformatics Pipelines | Genotypic analysis | Comprehensive resistance gene detection | ResFinder/PointFinder for E. coli WGS data [69] |
| Genetic Algorithm-AutoML Hybrid | Transcriptomic prediction | Identifies minimal predictive gene sets | 35-40 gene classifiers for P. aeruginosa [72] |
| RT-PCR Reagents | Expression validation | Quantifies resistance gene expression | Verification of silent resistance genes [32] |
Discussion and Future Directions
Genotype-phenotype discordance in resistance testing stems from multiple biological and technical factors. For Gram-positive organisms, where single mechanisms often confer resistance (e.g., mecA for methicillin resistance in S. aureus), prediction accuracy reaches 98-100% [32]. In contrast, Gram-negative organisms present greater complexity due to heterogeneous resistance mechanisms including porin loss, efflux pump overexpression, and undetected β-lactamase variants [32]. Additionally, transcriptomic studies reveal that resistance acquisition associates with changes in diverse regulatory and metabolic genes beyond canonical resistance markers [72].
Emerging approaches address these challenges through several strategies: Machine learning applied to transcriptomic data identifies minimal, predictive gene sets (35-40 genes) achieving 96-99% accuracy for P. aeruginosa resistance prediction [72]. These classifiers utilize genetic algorithms to select features from thousands of possibilities, often identifying previously unannotated genes beyond Comprehensive Antibiotic Resistance Database (CARD) annotations [72]. Functional prediction from 16S rRNA data, however, shows limited utility for clinical AMR prediction, with F1 scores of 0.08-0.22 for carbapenem resistance in E. coli [71].
The integration of rapid molecular detection with conventional phenotypic testing remains essential, particularly for complex cases. Standardized protocols for investigating discordant results help maintain diagnostic accuracy while advancing our understanding of resistance mechanisms. As databases expand and analytical methods improve, genotype-based prediction will likely play an increasingly prominent role in resistance monitoring and therapeutic guidance.
Optimizing Sample Preparation for Complex Matrices
In the field of antimicrobial resistance (AMR) research, the dichotomy between phenotypic and genotypic testing presents a significant challenge. Phenotypic resistance describes the observable ability of a bacterial population to survive or multiply in the presence of an antibiotic, typically measured through minimum inhibitory concentration (MIC) assays [5]. In contrast, genotypic resistance refers to the identification of specific genetic determinants—such as resistance genes or mutations—that confer the potential for resistance, regardless of whether this potential is currently expressed [5]. While phenotypic testing remains the cornerstone for determining appropriate antibiotic therapy, its major limitation is the prolonged incubation time required, which can critically delay targeted treatment decisions [73] [32]. This delay contributes to inappropriate empirical therapy, worsened patient prognoses, and the broader challenge of antimicrobial stewardship [73].
Sample preparation serves as the critical bridge between a complex clinical sample and a reliable analytical result. The presence of inhibitory substances in biological matrices, low abundance of target analytes, and the requirement for viable bacterial cells for phenotypic characterization make sample preparation a formidable bottleneck [74] [75]. Efficient and rapid preparation of samples is therefore not merely a technical step, but a fundamental prerequisite for accelerating AMR research and diagnostics. This application note provides detailed protocols and data for optimizing sample preparation, specifically within the context of resolving phenotypic and genotypic resistance profiles for faster therapeutic interventions.
Sample Preparation Strategies for Downstream Resistance Analysis
The choice of sample preparation method directly influences the sensitivity, specificity, and turnaround time of downstream phenotypic and genotypic analyses. The following section outlines optimized protocols for different analytical endpoints.
Protocol 1: Rapid Bacterial Isolation from Whole Blood for Culture-Free Analysis
Traditional blood culture systems, while sensitive, introduce significant delays. This protocol enables the direct isolation of bacterial pathogens from whole blood within 30 minutes, facilitating rapid identification via MALDI-TOF MS or molecular methods without the need for prior culture [74].
- 1. Objective: To efficiently isolate viable bacterial cells directly from human blood samples, minimizing diagnostic delays.
- 2. Applications: Downstream MALDI-TOF MS identification; genomic DNA extraction for PCR-based resistance gene detection; rapid phenotypic susceptibility testing from isolated cells.
- 3. Materials & Reagents:
- Anticoagulated Whole Blood Sample (e.g., collected in EDTA or heparin tubes).
- Red Blood Cell (RBC) Lysis Buffer: Ammonium chloride-based, sterile-filtered.
- Centrifugation Tubes (15 mL or 50 mL conical tubes).
- Phosphate-Buffered Saline (PBS), sterile and ice-cold.
- Benchtop Centrifuge capable of cooling to 4°C.
- Sterile Filter Unit (0.22 µm pore size) for buffer sterilization.
- 4. Procedure:
- Sample Mixing: Gently invert the blood collection tube 8-10 times to ensure homogeneity.
- RBC Lysis: Transfer 1 mL of whole blood to a 15 mL centrifuge tube. Add 10 mL of ice-cold RBC lysis buffer. Mix thoroughly by inverting the tube 10-15 times.
- Incubation: Incubate the mixture on ice for 10 minutes, inverting periodically to maintain lysis.
- Pelletting: Centrifuge at 4,000 x g for 10 minutes at 4°C to pellet the bacterial cells and white blood cells.
- Supernatant Removal: Carefully decant and discard the supernatant.
- Wash: Resuspend the pellet in 10 mL of ice-cold PBS. Centrifuge again at 4,000 x g for 10 minutes at 4°C.
- Final Resuspension: Discard the supernatant and resuspend the final pellet in 100 µL of PBS suitable for downstream analysis.
- 5. Critical Notes:
- Efficiency: This protocol achieves over 70% bacterial isolation efficiency and remains effective at low bacterial concentrations (1–10 CFU/0.3 mL blood) [74].
- Viability: The protocol preserves bacterial viability, with no notable change in growth lag times, allowing for subsequent culture-based confirmation if needed [74].
- Speed: The entire process is completed within 30 minutes, drastically reducing the time to result compared to standard blood culture [74].
Protocol 2: Optimized MALDI Matrix Preparation for Oligonucleotide and Biomolecular Analysis
Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) Mass Spectrometry is a powerful tool for rapid pathogen identification. However, its application to oligonucleotides (e.g., for resistance plasmid analysis) or other small molecules is hampered by low ionization efficiency and adduct formation. This protocol details the preparation of an optimized ionic matrix to enhance analytical performance [76].
- 1. Objective: To prepare a homogeneous ionic matrix that improves ionization efficiency, reduces alkali metal adducts, and enhances signal reproducibility for oligonucleotide analysis via MALDI-TOF MS.
- 2. Applications: Analysis of oligonucleotides; detection of biomolecules from complex mixtures; direct analysis from minimally processed samples.
- 3. Materials & Reagents:
- Matrix Compound: 6-Aza-2-thiothymine (ATT), purity ≥ 98%.
- Organic Base: 1-Methylimidazole (1-MI), purity 99%.
- Solvent: Methanol (MeOH), analytical grade.
- Additive Solution: Diammonium hydrogen citrate (DAC), 10 mg mL⁻¹ in ACN/H₂O (1:1, vol/vol).
- Vortex Mixer.
- SpeedVac or Nitrogen Evaporation System.
- MALDI Target Plate.
- 4. Procedure:
- Ionic Salt Formation: Dissolve ATT in MeOH at a concentration of 20 mg mL⁻¹. Add an equimolar amount of 1-MI to the solution.
- Mixing: Vortex the mixture for 5 minutes until fully dissolved.
- Drying: Evaporate the solvent to dryness using a SpeedVac or under a gentle stream of nitrogen.
- Reconstitution: Reconstitute the dried ionic matrix salt in the DAC additive solution to a final concentration of 75 mg mL⁻¹.
- Spotting (Two-Layer Method):
- Apply 0.5 µL of the matrix solution to the MALDI target and allow it to dry completely at room temperature.
- Subsequently, deposit 0.5 µL of the purified sample (e.g., extracted oligonucleotides) onto the pre-spotted matrix crystal layer and allow to dry [76].
- 5. Critical Notes:
- Performance: The ATT/1-MI ionic matrix with DAC additive consistently results in reduced standard deviation of mass-to-charge ratios and achieves high mass precision [76].
- Homogeneity: Ionic matrices form more homogeneous spots, leading to improved spectrum reproducibility and enhanced sensitivity [76].
- Adduct Suppression: The addition of DAC helps suppress the formation of sodium and potassium adducts, which is a major challenge in oligonucleotide analysis [76].
Research Reagent Solutions
The table below summarizes key reagents and their critical functions in the sample preparation workflows described in this note.
Table 1: Essential Research Reagents for Sample Preparation in Resistance Testing
| Reagent | Function/Application | Key Property / Consideration |
|---|---|---|
| 6-Aza-2-thiothymine (ATT) [76] | Ionic matrix for MALDI-TOF MS | Enhances ionization efficiency and spot homogeneity for oligonucleotides and other biomolecules. |
| 1-Methylimidazole (1-MI) [76] | Organic base for ionic matrix formation | Forms a stable organic salt with conventional matrices, improving reproducibility. |
| Diammonium Hydrogen Citrate (DAC) [76] | Additive for MALDI matrix | Suppresses alkali metal adduct formation, leading to cleaner spectra and higher resolution. |
| Ammonium Chloride Lysis Buffer [74] | Rapid RBC lysis | Selectively lyses red blood cells while preserving the integrity of bacterial pathogens for isolation. |
| ACN/H₂O Solvent Mixture [76] | Solvent for matrix and additive preparation | Standard solvent system that ensures proper dissolution and crystallization of MALDI matrices. |
Data Presentation and Analysis
The correlation between optimized sample preparation and analytical output is quantifiable. Systematic evaluation of 48 samples prepared with different matrices for oligonucleotide analysis revealed that only 19 met the signal-to-noise (S/N) criteria for detection across a mass range of 4–10 kDa, underscoring the critical role of matrix selection [76].
Table 2: Impact of Sample Preparation on MALDI-TOF MS Analytical Performance for Oligonucleotides
| Matrix Formulation | Key Additive | Signal-to-Noise (S/N) Ratio | Mass Precision | Key Finding |
|---|---|---|---|---|
| ATT + 1-MI (Ionic Matrix) [76] | 1-Methylimidazole | Consistently High | High | Reduced standard deviation; achieved high mass precision. |
| 3-Hydroxypicolinic Acid (3-HPA) [76] | DAC | Variable | Variable | Performance highly dependent on solvent composition and additives. |
| 2,4,6-THAP [76] | DAC | Not Specified | Not Specified | DAC additive suppresses alkali ion adducts, increasing signal intensity. |
| ATT or 3-HPA [76] | Fucose | Improved | Not Specified | Additive led to reduced fragmentation and increased spot homogeneity. |
Workflow and Pathway Diagrams
The following diagram illustrates the logical workflow for selecting a sample preparation strategy based on the intended downstream application in phenotypic vs. genotypic resistance research.
Diagram 1: Sample preparation workflow for resistance testing.
Optimizing sample preparation is a decisive factor in successfully navigating the complexities of phenotypic and genotypic resistance testing. As demonstrated, tailored protocols can dramatically reduce the time from sample to result, directly addressing a key limitation of phenotypic AST [74]. Furthermore, refined preparation methods for mass spectrometry unlock its potential for broader applications, including the detection of resistance-associated oligonucleotides, thereby enriching genotypic insights [76].
The interplay between phenotype and genotype is not always straightforward. While the detection of a resistance gene (genotype) often predicts resistance (phenotype), discrepancies are common. For instance, in Gram-negative bacteria, the absence of a detected resistance gene (e.g., blaKPC) does not guarantee susceptibility, as off-panel mechanisms or efflux pumps may confer resistance [32]. Therefore, robust sample preparation that supports both phenotypic and genotypic workflows provides a comprehensive diagnostic and research strategy. The protocols outlined here provide a foundation for developing integrated, rapid testing pipelines. By improving the quality and speed of the initial analytical step, researchers and clinicians can generate more reliable data faster, ultimately contributing to more timely and effective patient therapy and bolstered antimicrobial stewardship efforts.
Table 1: Comparative Performance and Market Analysis of AMR Diagnostic Technologies
| Parameter | Microbiology Culture (Phenotypic) | PCR (Genotypic) | NGS (Genotypic) | Mass Spectrometry | Rapid & Point-of-Care |
|---|---|---|---|---|---|
| Typical Turnaround Time | 24 - 72 hours [77] [62] | A few hours [77] | Varies (longer than PCR) | N/A | Under 1 hour (e.g., for UTI) [77] |
| Key Strength | Functional assessment of resistance, gold standard | High sensitivity and speed for known genes [77] [78] | Comprehensive detection of resistance mechanisms [77] | N/A | Rapid results for clinical decision-making [77] [78] |
| Key Limitation | Slow turnaround time [77] | Limited to pre-defined targets | High cost, complex data analysis [78] | N/A | Limited multiplexing capability |
| Market Share (Technology) | N/A | Dominant segment [77] [78] | Growing segment | 14% [79] | Growing segment [78] |
| Projected Market CAGR | N/A | N/A | N/A | N/A | N/A |
| Primary Cost Driver | Labor, consumables | Instrumentation, specialized reagents [79] | Instrumentation, bioinformatics | High capital investment [79] | Test cartridge/disposables |
Table 2: Global Market Metrics and Regional Growth for AMR Diagnostics
| Market Segment | Value (2024/2025) | Projected Value (2032/2035) | Projected CAGR | Key Growth Region |
|---|---|---|---|---|
| Overall AMR Diagnostics Market | US$ 4.5 Bn (2024) [78] | US$ 8.9 Bn (2035) [78] | 6.5% [78] | North America [77] [78] |
| Overall AMR Diagnostics Market | US$ 4,830.7 Mn (2025) [77] | US$ 7,620.1 Mn (2032) [77] | 6.7% [77] | Asia-Pacific (Highest Growth) [77] |
| Antibiotic Susceptibility Testing (AST) Devices Market | USD 361.1 Mn (2025) [79] | USD 599.5 Mn (2035) [79] | 5.2% [79] | East Asia, South Asia Pacific [79] |
| Reagents & Consumables Segment | Dominant product segment [77] | N/A | N/A | Global |
Experimental Protocols
Protocol 1: Phenotypic Detection of Metallo-β-Lactamase (MBL) Production inAcinetobacter baumannii
This protocol outlines the methodology for the phenotypic identification of MBL production in drug-resistant A. baumannii isolates, as used in a recent clinical study [62].
Materials and Reagents
- Mueller Hinton Agar (MHA) Plates
- Antibiotic Impregnated Disks: Imipenem (10 µg) and Meropenem (10 µg)
- MBL Inhibitor Solution: 0.5 M Ethylenediaminetetraacetic acid (EDTA)
- Sterile Saline Solution (0.85%)
- McFarland Standard: 0.5
- Sterile Cotton Swabs
Procedure: Double-Disk Synergy Test (DDST)
- Preparation of Inoculum: Adjust the turbidity of a fresh, pure bacterial suspension in sterile saline to match the 0.5 McFarland standard.
- Inoculation: Using a sterile swab, evenly spread the inoculum over the entire surface of a MHA plate to create a uniform lawn culture.
- Disk Placement:
- Aseptically place an imipenem or meropenem disk on the inoculated agar.
- Aseptically place a disk containing 10 µL of 0.5 M EDTA at a distance of 15 mm center-to-center from the antibiotic disk.
- Incubation: Invert the plate and incubate aerobically at 35°± 2°C for 16-18 hours.
- Interpretation: A positive result for MBL production is indicated by an enhanced zone of inhibition between the antibiotic disk and the EDTA disk, forming a "keyhole" or synergy zone [62].
Protocol 2: Genotypic Profiling of Antimicrobial Resistance and Virulence Factors inPseudomonas aeruginosa
This protocol describes a comprehensive genotypic analysis workflow for identifying acquired resistance genes and virulence factors in P. aeruginosa isolates from keratitis patients using whole-genome sequencing (WGS) data [63].
Materials and Reagents
- Genomic DNA from bacterial isolates (e.g., from 70 corneal P. aeruginosa isolates)
- Bioinformatics Software/Tools:
- Prokka v1.14.6: For rapid annotation of prokaryotic genomes.
- ResFinder: For identification of acquired antimicrobial resistance genes.
- Virulence Factor Database (VFDB): For screening virulence-associated genes.
- Roary v3.13.0: For pangenome analysis.
- Snippy v4.6.0: For whole-genome variant calling.
- MobileElementFinder v1.0.3 & IslandViewer 4: For identifying mobile genetic elements and pathogenicity islands.
- Computing Infrastructure: High-performance computing cluster or server with adequate RAM and processing power.
Procedure: Genomic Analysis Workflow
- Data Acquisition and Annotation:
- Retrieve raw WGS data from a database such as the National Center for Biotechnology Information (NCBI).
- Annotate the genomes using Prokka to identify all coding sequences.
- Resistance and Virulence Profiling:
- Analyze the annotated genomes against the Comprehensive Antibiotic Resistance Database (CARD) using ResFinder to determine the AMR gene profile.
- Screen the genomes against the Virulence Factor Database (VFDB) to identify virulence factors.
- Comparative Genomics:
- Perform a pangenome analysis using Roary to identify core, accessory, and unique genes among the isolates.
- Use Snippy for variant analysis to identify single nucleotide polymorphisms (SNPs) and insertions/deletions (indels).
- Horizontal Gene Transfer Analysis:
- Identify mobile genetic elements (e.g., insertion sequences, transposons) using MobileElementFinder.
- Predict genomic islands associated with pathogenicity using IslandViewer 4.
Workflow Visualization
AMR Testing Strategy Selection
Genotypic AMR Analysis Pipeline
Research Reagent Solutions
Table 3: Essential Research Tools for AMR Diagnostics Development
| Reagent / Material | Function / Application | Example Use-Case / Note |
|---|---|---|
| Mueller Hinton Agar (MHA) | Standardized medium for antimicrobial susceptibility testing (AST) by disk diffusion. | Used in the Double-Disk Synergy Test for phenotypic MBL detection [62]. |
| MBL-E Test Strips | Quantitative phenotypic test for Metallo-β-Lactamase production. | Used as a comparative method in studies validating genotypic assays [62]. |
| VITEK 2 AST Cards | Automated, miniaturized broth dilution system for rapid AST. | FDA-cleared system; part of integrated automated solutions [77] [80]. |
| Selux AST System | Next-generation automated phenotypic susceptibility testing system. | Cleared by FDA with panels that can expand to incorporate new drugs [80]. |
| Helini Biomolecules Kits | Reagents for molecular detection of resistance genes (e.g., blaNDM, blaOXA-58, blaVIM). | Used in clinical studies for genotypic MBL confirmation [62]. |
| Prokka Software | Rapid, automated annotation of prokaryotic genomes. | Used in WGS-based studies to annotate bacterial genomes prior to AMR gene analysis [63]. |
| ResFinder / CARD | Bioinformatics tools for identifying acquired antibiotic resistance genes from genomic data. | Critical for genotypic profiling in research studies using WGS data [63]. |
The shift from phenotypic to genotypic antimicrobial resistance (AMR) detection, propelled by whole genome sequencing (WGS), represents a paradigm change in clinical microbiology [81]. While genotypic predictions promise speed and comprehensiveness, their clinical utility is constrained by significant interpretation complexities. The accuracy of WGS-based antimicrobial susceptibility testing (WGS-AST) is highly dependent on the bioinformatics pipelines and resistance databases used for analysis [81] [82]. Discrepancies between genotypic predictions and phenotypic results highlight critical knowledge gaps in our understanding of resistance mechanisms and the limitations of current computational tools. This application note examines these complexities within the broader context of phenotypic versus genotypic resistance testing, providing structured data, validated protocols, and visualization tools to enhance research reproducibility and clinical decision-making.
Quantitative Comparison of Pipeline and Database Performance
Concordance Between Phenotypic and Genotypic Drug Susceptibility Testing
Table 1: Agreement between phenotypic and genotypic DST for M. tuberculosis isolates (n=63) [83]
| Anti-TB Drug | Concordance Rate (%) | Level of Agreement |
|---|---|---|
| Isoniazid | 100% | Perfect |
| Rifampicin | 100% | Perfect |
| Linezolid | 100% | Perfect |
| Ofloxacin/Levofloxacin | 93.7% | High |
| Pyrazinamide | 93.7% | High |
| Streptomycin | 95.4% | High |
| Ethambutol | 85.7% | Moderate |
| Amikacin | 82.5% | Moderate |
| Kanamycin | 85.4% | Moderate |
| Capreomycin | 81.0% | Moderate |
| Moxifloxacin | 77.8% | Moderate |
| Ethionamide | 56.4% | Poor |
Performance Metrics of Bioinformatics Tools for AMR Gene Identification
Table 2: Pipeline validation metrics for AMR gene identification in K. pneumoniae (n=201 genomes) [84]
| Validation Metric | ABRicate Performance | ResFinder Performance |
|---|---|---|
| Repeatability | 100% | 100% |
| Reproducibility | 100% | 100% |
| Average Number of AMR Genes Identified | 15.85 ± 0.39 | 23.27 ± 0.56 |
| Gene Duplication Incidence | 8 samples | All samples (up to 6×) |
| Coverage Percentage (All Genes) | Higher (p < 0.0001) | Lower |
| Identity Percentage (All Genes) | Higher (p = 0.0002) | Lower |
| Recommended for AMR Gene Identification | Yes | No |
Experimental Protocols
Protocol 1: Whole Genome Sequencing and Analysis of Mycobacterium tuberculosis Isolates
Principle: This protocol describes the methodology for comparing phenotypic drug susceptibility testing (DST) with genotypic predictions derived from whole genome sequencing of M. tuberculosis isolates, specifically targeting multidrug-resistant strains [83].
Materials:
- Clinical M. tuberculosis isolates
- BACTEC MGIT 960 system and/or Lowenstein-Jensen solid media
- GenoLyse DNA extraction kit (Hain Lifescience, Germany)
- Nucleomag magnetic beads (Macherey-Nagel, Netherlands)
- QIAseq FX DNA library kit (Qiagen, Germany)
- MiSeq platform (Illumina, US)
Procedure:
- Sample Selection and Phenotypic DST:
- Select MDR-TB isolates (resistant to at least isoniazid and rifampicin) with available additional DST data.
- Perform phenotypic DST using BACTEC MGIT 960 system and/or Lowenstein-Jensen solid media according to WHO technical manual [83].
- Test against a panel of anti-TB drugs including isoniazid, rifampicin, ethambutol, pyrazinamide, streptomycin, amikacin, kanamycin, capreomycin, ofloxacin, moxifloxacin, ethionamide, and linezolid.
DNA Extraction and Purification:
- Extract bacterial DNA using GenoLyse kit according to manufacturer's protocol.
- Purify DNA samples using Nucleomag magnetic beads in a 1:2 ratio.
- Elute DNA in 10 mM Tris buffer solution (pH = 8).
Library Preparation and Sequencing:
- Prepare paired-end fragment libraries using QIAseq FX DNA library kit.
- Sequence on MiSeq platform (Illumina) producing reads of maximum 600 bp.
Bioinformatic Analysis:
- Process sequencing data on Galaxy web platform.
- Trim adapter sequences and low-quality ends using Trimmomatic (v0.38.0) keeping sequences of at least 20 base pairs long.
- Map outputs to reference sequence (H37Rv, GenBank NC000962.3) using snippy tool (v4.6.0).
- Perform left aligning of indels using BamLeftAlign (v1.3.6).
- Filter BAM files to keep only properly paired mapped reads and remove PCR duplicates.
- Analyze generated BAM file using TB-Profiler tool (v4.1.1) to discriminate lineages and detect resistance-associated mutations.
- Manually check detected changes in resistance-related genes against WHO mutation catalogue.
- Call mutations with allele frequency ≥ 10% supported by at least four sequencing reads.
Genotypic DST Interpretation:
- Assume "Resistant" genotypic DST result if group 1 ("Associated with resistance"), group 2 ("Associated with resistance, interim"), or group 3 ("Uncertain significance") mutations are detected.
- Assume "Sensitive" genotypic DST result if group 4 ("Not associated with resistance, interim"), group 5 ("Not associated with resistance"), or non-graded mutations are detected.
Concordance Analysis:
- Compare phenotypic and genotypic DST results for each drug.
- Calculate concordance rates as percentage agreement between methods.
Calculation:
Protocol 2: Bioinformatics Pipeline Validation for Antimicrobial Resistance Gene Identification
Principle: This protocol validates a bioinformatics pipeline for in silico analysis of WGS data from carbapenem-resistant K. pneumoniae isolates to produce standardized data enabling interlaboratory comparisons [84].
Materials:
- K. pneumoniae whole genome sequences from public databases (NCBI)
- Computational resources for bioinformatic analysis
- Kraken2 for bacterial identification
- SpeciesFinder for bacterial identification
- ResFinder for AMR gene identification
- ABRicate for AMR gene identification
Procedure:
- Data Acquisition and Quality Control:
- Obtain K. pneumoniae genome sequences from public databases such as BV-BRC or NCBI.
- Exclude outlier genomes with >250 contigs and lengths >6.4 Mbp or <4.9 Mbp.
- Perform species typing using Kleborate v2.2.0 to remove non-K. pneumoniae samples.
Genome Assembly and Annotation:
- Trim raw sequencing reads to remove adapters and low-quality sequences.
- Perform de novo assembly of trimmed reads.
- Order assembled contigs against a reference genome.
- Annotate assembled genomes for gene content.
Bacterial Identification:
- Analyze all samples using Kraken2 and SpeciesFinder tools.
- Compare identification results between tools.
- Calculate reproducibility and repeatability of identification.
AMR Gene Identification:
- Annotate samples using ResFinder and ABRicate with default settings.
- Compile list of identified AMR genes, focusing on carbapenem resistance genes (blaKPC, blaNDM, blaOXA, blaVIM).
- Compare number of AMR genes identified by each tool.
- Note instances of gene duplication in results.
Validation Metrics Calculation:
- Analyze sequences in triplicate on the same day to determine repeatability.
- Evaluate samples with technical replicates on alternate days to calculate reproducibility.
- Compare results with reference sequences to calculate accuracy, precision, sensitivity, and specificity.
Calculations:
- Parameter Sensitivity Analysis:
- Identify AMR genes using ABRicate with parameters set at 90% identity and 60% coverage (ResFinder defaults).
- Identify AMR genes using ResFinder with parameters set at 80% identity and coverage (ABRicate defaults).
- Compare results under different parameter settings.
Visualization of Workflows and Relationships
Bioinformatics Pipeline for AMR Analysis
Bioinformatics AMR Analysis Pipeline
Phenotypic vs Genotypic Correlation Analysis
Phenotypic vs Genotypic Correlation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential materials and bioinformatics tools for AMR research
| Category | Item | Function/Application |
|---|---|---|
| Wet Lab Materials | GenoLyse DNA Extraction Kit | DNA extraction from mycobacterial isolates [83] |
| BACTEC MGIT 960 System | Automated phenotypic DST for mycobacteria [83] | |
| Lowenstein-Jensen Solid Media | Solid media for phenotypic DST [83] | |
| QIAseq FX DNA Library Kit | Library preparation for WGS [83] | |
| MiSeq Platform (Illumina) | Whole genome sequencing [83] | |
| Bioinformatics Tools | TB-Profiler | Lineage discrimination and resistance mutation detection for M. tuberculosis [83] |
| ResFinder | Identification of acquired antimicrobial resistance genes [84] | |
| ABRicate | AMR gene detection with CARD database [84] | |
| AMRFinderPlus | Comprehensive AMR gene and mutation detection [82] | |
| Kraken2 | Rapid metagenomic sequence classification [84] | |
| Kleborate | K. pneumoniae-specific genotyping and resistance profiling [82] | |
| Databases | CARD (Comprehensive Antibiotic Resistance Database) | Curated resource of resistance genes, mutations, and mechanisms [81] |
| WHO Mutation Catalogue | Confidence-graded Mtb genetic markers for drug resistance [83] | |
| ResFinder Database | Database of resistance genes for bacterial pathogens [82] |
The escalating global threat of antimicrobial resistance (AMR) necessitates robust laboratory methods to accurately characterize resistance mechanisms. The dichotomy between phenotypic testing, which measures the observable growth response of microbes to antibiotics, and genotypic testing, which detects specific resistance genes or mutations, forms a central theme in modern microbiological research and clinical diagnostics [85] [86]. While genotypic methods offer speed, phenotypic assays provide the functional context essential for understanding intrinsic resistance—the innate ability of a microbe to resist an antibiotic class due to its structural or genetic makeup [15] [24]. This application note details standardized protocols for both methodological approaches, framed within quality assurance principles to ensure reproducibility, reliability, and translational relevance in resistance research.
Comparative Analysis of Resistance Testing Methodologies
The choice between phenotypic and genotypic testing is not mutually exclusive but rather complementary. Each approach offers distinct advantages and addresses different stages of the research and diagnostic pipeline. The following table summarizes the core characteristics of each method.
Table 1: Core Characteristics of Phenotypic and Genotypic Resistance Testing Methods
| Feature | Phenotypic Testing | Genotypic Testing |
|---|---|---|
| Fundamental Principle | Measures direct effect on microbial growth/replication in the presence of an antibiotic [87] [88]. | Detects specific genetic sequences (genes, mutations) known to confer resistance [62] [87]. |
| Key Advantage | Functional, comprehensive readout of resistance; detects novel/uncharacterized mechanisms [85] [86]. | High speed, specificity, and predictive value; identifies resistance before it is phenotypically expressed. |
| Primary Limitation | Time-consuming (typically 18-24 hours); result is an observation without immediate mechanistic explanation [62]. | Requires prior knowledge of resistance determinants; may miss novel mechanisms or those not included in the assay panel [89]. |
| Typical Output | Minimum Inhibitory Concentration (MIC), zone of inhibition diameter, growth/no-growth determination [90]. | A list of detected mutations or acquired resistance genes compared to a reference sequence [87] [88]. |
| Ideal Application | Gold-standard for susceptibility profiling, confirming resistance phenotype, functional genomics screens [15] [24]. | Rapid screening, epidemiology, outbreak investigation, and guiding targeted therapy [62] [63]. |
Experimental Protocols for Intrinsic Resistance Research
Standardized protocols are the bedrock of quality assurance. The following sections provide detailed methodologies for key experiments in intrinsic resistance research.
Protocol 1: Phenotypic Screen for Intrinsic Resistance Genes
This protocol, adapted from a genome-wide screening approach, identifies bacterial genes that contribute to intrinsic antibiotic resistance when knocked out [15] [24].
I. Primary Screening in Liquid Culture
- Library Preparation: Obtain a comprehensive single-gene knockout library (e.g., the Keio collection for E. coli).
- Culture Setup: In a 96-well plate, inoculate each knockout strain in duplicate into two types of media:
- Test Medium: Luria-Bertani (LB) broth supplemented with the antibiotic of interest at a predetermined sub-inhibitory concentration (e.g., IC~50~).
- Control Medium: LB broth without antibiotic.
- Incubation and Measurement: Incubate plates with shaking at 37°C for a defined period (e.g., 16-20 hours). Measure the optical density at 600 nm (OD~600~) for each well.
- Data Analysis: For each knockout strain, calculate the growth in the test condition as a fraction of its growth in the control condition. Normalize this value to the wild-type strain's performance. Identify "hypersensitive" hits as those with growth significantly lower (e.g., >2 standard deviations below the median) than the population average [15].
II. Secondary Validation on Solid Media
- Spot Assay: From the primary hits, spot knockout strains onto agar plates supplemented with a range of antibiotic concentrations (e.g., MIC, MIC/3, MIC/9).
- Analysis: After incubation, score colony formation. Hypersensitive mutants will show compromised growth on antibiotic-supplemented agar compared to the wild-type control [15] [24].
Protocol 2: Genotypic Detection of Carbapenem Resistance Genes
This protocol outlines the molecular detection of carbapenemase genes (e.g., bla~NDM~, bla~OXA-48~, bla~VIM~) from bacterial isolates using polymerase chain reaction (PCR) [62].
- DNA Extraction: Purify genomic DNA from bacterial colonies using a commercial kit. Quantify DNA concentration and purity via spectrophotometry.
- Primer Preparation: Reconstitute and dilute lyophilized primers specific for the target resistance genes. The table below lists example primers.
Table 2: Example Primer Sequences for Carbapenemase Gene Detection [62]
| Target Gene | Primer Name | Sequence (5' to 3') |
|---|---|---|
| blaNDM | blaNDM-F | AACACAGCCTGACTTTCG |
| blaNDM-R | TGATATTGTCACTGGTGTGG | |
| blaOXA-58 | blaOXA-58-F | TGGCACGCATTTAGACCG |
| blaOXA-58-R | AAACCCACATACCAACCC | |
| blaVIM | blaVIM-F | GATGGTGTTTGGTCGCATA |
| blaVIM-R | CGAATGCGCAGCACCAG |
- PCR Setup: Prepare a reaction mix containing PCR master mix, forward and reverse primers for each target, nuclease-free water, and template DNA. Include positive and negative controls.
- Amplification: Run the PCR using a validated thermal cycling protocol. An example cycle is: initial denaturation at 95°C for 5 min; 35 cycles of 95°C for 30s, 55-60°C (primer-specific) for 30s, and 72°C for 1 min/kb; final extension at 72°C for 7 min.
- Amplicon Analysis: Analyze PCR products by gel electrophoresis. The presence of a band at the expected size confirms the detection of the target resistance gene.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of resistance research relies on a core set of reliable reagents and resources.
Table 3: Key Research Reagent Solutions for Resistance Studies
| Reagent / Resource | Function and Application | Example / Specification |
|---|---|---|
| Knockout Library | Enables genome-wide screening to identify genes involved in intrinsic resistance. | Keio collection (E. coli) [15] [24] |
| Mueller-Hinton Agar | The standardized medium for antibiotic susceptibility testing, ensuring reproducible results. | Prepared according to CLSI guidelines [90] |
| Antibiotic Discs/E-Strips | For phenotypic determination of resistance levels (MIC, zone of inhibition). | Commercially sourced, quality-controlled discs and MIC strips [62] [89] [90] |
| Primer Panels | Specific oligonucleotides for amplification and detection of known resistance genes. | Custom or commercial primers for genes like bla~NDM~, bla~OXA~, bla~VIM~ [62] |
| Positive Control Strains | Essential quality control for both phenotypic and genotypic assays. | NCTC-13304 (positive), NCTC-13302 (negative) [62] |
| Commercial Resistance Databases | Curated knowledge bases for interpreting genotypic findings and predicting phenotypes. | CARD, ResFinder [63] |
Visualizing Workflows and Resistance Pathways
Standardized workflows are critical for quality assurance. The following diagrams map the logical pathways for resistance testing and the mechanism of intrinsic resistance breakers.
Phenotypic and Genotypic Testing Workflow
This diagram illustrates the parallel and integrated paths of phenotypic and genotypic testing in a research or diagnostic setting.
Targeting Intrinsic Resistance Pathways
This diagram conceptualizes the strategy of impairing intrinsic resistance pathways, such as efflux pumps or cell membrane integrity, to re-sensitize bacteria to antibiotics [15] [24].
The fight against antimicrobial resistance demands unwavering commitment to quality and standardization in the laboratory. By implementing the detailed application notes and protocols outlined herein—from standardized phenotypic screens and genotypic detections to the use of curated reagent toolkits and visual workflows—researchers can significantly enhance the reproducibility and translational impact of their work. A rigorous, integrated approach that leverages the functional depth of phenotyping and the predictive speed of genotyping is paramount for unraveling the complexities of intrinsic resistance and developing the next generation of effective therapeutics.
Performance Assessment: Comparative Analysis and Validation Frameworks
In the critical field of antimicrobial resistance (AMR) research, the evaluation of diagnostic tests requires standardized metrics to ensure accuracy and clinical relevance. Categorical Agreement (CA), Essential Agreement (EA), and error rate analysis form the cornerstone of method validation in the comparative study of phenotypic and genotypic antimicrobial susceptibility testing (AST) [91]. As AMR continues to pose a major global health burden, responsible for an estimated 1.2 million deaths annually, the importance of reliable and rapid diagnostics has never been greater [91] [92].
These metrics provide a rigorous framework for benchmarking new technologies—from rapid phenotypic systems to genotypic resistance gene detection—against reference methods [14]. For researchers and drug development professionals, understanding and correctly applying these metrics is paramount for developing novel diagnostics and therapeutic strategies, ultimately contributing to effective antimicrobial stewardship and patient care.
Defining the Core Evaluation Metrics
The evaluation of any new AST method against a reference standard involves quantifying the level of concordance through specific, standardized metrics. The most critical of these are defined below.
Table 1: Definitions of Key AST Evaluation Metrics
| Metric | Definition | Interpretation in AST Context | Acceptance Criteria |
|---|---|---|---|
| Categorical Agreement (CA) | The percentage of isolates where the new test and reference method yield the same categorical interpretation (Susceptible, Intermediate, or Resistant). [93] [94] | Measures clinical interpretative agreement. | ≥ 90% [93] [94] |
| Essential Agreement (EA) | The percentage of isolates where the Minimum Inhibitory Concentration (MIC) obtained by the new test is within ±1 two-fold dilution of the reference MIC. [93] [94] | Measures quantitative MIC precision, independent of clinical breakpoints. | ≥ 90% [93] [94] |
| Very Major Error (VME) | The percentage of isolates that are resistant by the reference method but are categorized as susceptible by the new test. [93] [94] | Represents a false susceptible result; the most serious error as it could lead to treatment failure. | ≤ 1.5% - 3% [93] [94] |
| Major Error (ME) | The percentage of isolates that are susceptible by the reference method but are categorized as resistant by the new test. [93] [94] | Represents a false resistant result; may lead to the unnecessary avoidance of an effective drug. | ≤ 3% [93] [94] |
| Minor Error (mE) | The percentage of isolates where the new test and reference method result in differing interpretations that are both either susceptible or resistant (e.g., S to I, I to R, or vice-versa). [93] | An error that may affect reporting but has less immediate clinical impact than VME or ME. | ≤ 10% [93] |
The Critical Role of Error Rates
Error rates, particularly VME and ME, carry significant clinical implications. A Very Major Error (VME) is considered the most critical failure, as it misclassifies a resistant isolate as susceptible, potentially leading to the administration of an ineffective antibiotic and subsequent treatment failure [93] [94]. Conversely, a Major Error (ME) could cause a clinician to reject a viable therapeutic option, potentially leading to the use of a broader-spectrum antibiotic than necessary [94]. The stringent acceptance criteria for these metrics (e.g., VME ≤ 1.5%) underscore their importance in ensuring patient safety and effective therapy.
Application in Comparative AST Research
The metrics of CA, EA, and error rates are universally applied across AST development, from evaluating novel phenotypic methods to assessing the correlation between genotypic and phenotypic results.
Evaluating Novel Phenotypic Methods
Recent studies on rapid phenotypic platforms consistently report these metrics to validate their performance. For instance, a study comparing the E-test and broth microdilution (BMD) for testing polymyxin B against carbapenem-resistant Enterobacteriaceae (CRE) reported a CA of 99.13%, an EA of 95.65%, and a VME of 0.87%, demonstrating strong agreement between the methods. [93]
Another study developed a resazurin-based microdilution method for Neisseria gonorrhoeae and evaluated it against the agar dilution gold standard. The results, summarized in the table below, showed high EA but also highlighted areas requiring optimization, as indicated by the ME rates for some drugs. [94]
Table 2: Performance of Resazurin-Based Microdilution Method for N. gonorrhoeae [94]
| Antibiotic | Essential Agreement (EA) | Categorical Agreement (CA) | Very Major Error (VME) | Major Error (ME) |
|---|---|---|---|---|
| Azithromycin | 97.1% | 88.6% | 0% | 11.4% |
| Ceftriaxone | 91.5% | 94.3% | 0% | 5.7% |
| Spectinomycin | 94.3% | 94.3% | 0% | 5.7% |
Bridging Genotypic and Phenotypic Resistance
The correlation between genotype and phenotype is a central theme in AMR research. A study on Pasteurella multocida utilized WGS to identify resistance genes (ARGs) and compared the genotypic predictions with phenotypic results from disk diffusion and BMD. The findings indicated that "MIC values showed a stronger positive correlation with genotypic results" than disk diffusion, affirming BMD as the more reliable phenotypic reference method for such comparisons. [11] This type of analysis is crucial for validating genotypic predictions and understanding the complex relationship between the presence of a resistance gene and its phenotypic expression.
Experimental Protocols for Metric Calculation
To ensure the reproducibility and validity of AST evaluations, a standardized experimental protocol is essential. The following provides a detailed methodology for a comparative study, adaptable for various sample types and technologies.
Protocol: Broth Microdilution (Reference Method) for Gram-Negative Bacteria
1. Principle: The BMD method determines the Minimum Inhibricular Concentration (MIC) by visually assessing the inhibition of bacterial growth in a liquid medium containing serial dilutions of an antimicrobial agent. [93] [94]
2. Reagents and Materials:
- Cation-adjusted Mueller-Hinton Broth (CAMHB)
- Sterile, 96-well U-bottom microtiter plates
- Antimicrobial stock solutions
- Bacterial isolates (adjusted to 0.5 McFarland standard, then diluted to ~5x10^5 CFU/mL in broth) [94]
- Multichannel pipettes and sterile reservoirs
- Incubator (35±2°C)
3. Procedure: Step 1: Plate Preparation. Create serial two-fold dilutions of the antimicrobial agent in CAMHB directly in the microtiter plate. The final volume in each well should be 100 µL. Step 2: Inoculation. Add 100 µL of the prepared bacterial inoculum (~5x10^5 CFU/mL) to each well of the plate containing the antimicrobial dilutions. This achieves a final testing concentration of ~5x10^4 CFU/mL per well. Step 3: Controls. Include growth control wells (inoculum without antibiotic) and sterility controls (broth only). Step 4: Incubation. Incubate the plates for 16-24 hours at 35±2°C under ambient atmosphere. Step 5: MIC Reading. Read the MIC visually as the lowest concentration of antimicrobial agent that completely inhibits visible growth. [93] [94]
Protocol: Evaluating a Novel Resazurin-Based AST Method
1. Principle: This method uses the oxidation-reduction indicator resazurin, which changes from blue to pink/colorless in the presence of metabolically active bacteria, allowing for a faster, colorimetric endpoint. [94]
2. Reagents and Materials:
- All materials from the BMD protocol
- Sterile 0.01% (w/v) resazurin sodium salt solution
3. Procedure: Step 1: Perform BMD. Set up the BMD test as described in section 4.1. Step 2: Add Resazurin. After the initial incubation period (or at the time of inoculation for fast-growing organisms), add 50 µL of the sterile 0.01% resazurin solution to each well. Step 3: Secondary Incubation. Continue incubation for a predetermined, optimized period (e.g., 2-6 hours). Step 4: Colorimetric Reading. Read the MIC as the lowest antibiotic concentration where the well remains blue (no color change). A change to pink or colorless indicates bacterial growth and metabolic activity. [94]
4. Data Analysis:
- Calculate the EA by determining the percentage of isolates where the resazurin-based MIC is within ±1 doubling dilution of the BMD MIC.
- Interpret both the BMD and resazurin MICs using established clinical breakpoints (e.g., EUCAST). Calculate the CA, VME, and ME by comparing the categorical results from the two methods. [94]
Diagram 1: AST method evaluation workflow showing how metrics are calculated from phenotypic and novel test data.
The Scientist's Toolkit: Research Reagent Solutions
Successful AST research relies on a suite of critical reagents and tools. The following table details key materials and their functions in a standard evaluation workflow.
Table 3: Essential Research Reagents and Materials for AST Evaluation
| Item | Function/Application | Example Use in Protocol |
|---|---|---|
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized medium for broth microdilution AST; ensures consistent cation concentrations for reliable antibiotic activity. [93] | Used as the base medium for preparing antibiotic dilutions and bacterial inoculum in the reference BMD method. |
| 96-Well U-Bottom Microtiter Plates | Platform for housing serial antibiotic dilutions and bacterial inoculum in a high-throughput format. | The physical vessel for performing the broth microdilution test. |
| Antibiotic Reference Powder | High-purity, characterized powder for preparing in-house stock solutions of antimicrobial agents at precise concentrations. | Used to create the serial two-fold dilutions for MIC determination. |
| Resazurin Sodium Salt | Oxidation-reduction indicator; used in colorimetric AST methods to detect metabolic activity of viable bacteria. [94] | Added to microdilution wells to reduce time-to-result; a color change indicates bacterial growth. |
| Densitometer | Instrument to standardize bacterial inoculum density by measuring turbidity against McFarland standards. | Used to adjust the bacterial suspension to 0.5 McFarland standard prior to dilution for inoculation. |
| Clinical & Laboratory Standards Institute (CLSI) or EUCAST Guidelines | Documents providing standardized methodologies, quality control ranges, and clinical breakpoints for AST. [93] | Essential reference for ensuring the experimental protocol, quality control, and result interpretation are performed according to international standards. |
The escalating global antimicrobial resistance (AMR) crisis necessitates rapid, accurate diagnostic methods to guide effective patient therapy and stewardship efforts [95] [14]. Clinical microbiology laboratories primarily utilize two approaches for AMR detection: phenotypic methods, which measure observable microbial growth in the presence of antimicrobials, and genotypic methods, which identify specific resistance genes or mutations through molecular techniques [12] [95]. This application note provides a structured comparison of these methodologies, supported by quantitative performance data from recent studies, detailed experimental protocols for essential assays, and decision-support tools for implementation within a clinical research framework. The content is framed within a broader thesis investigating the intrinsic advantages and limitations of each approach, aiming to optimize their application and integration in both routine diagnostic and research settings.
Comparative Performance Data
Recent comparative studies across diverse bacterial pathogens highlight the operational characteristics and diagnostic performance of phenotypic versus genotypic susceptibility testing methods. The data below summarize key findings from evaluations conducted in 2024-2025.
Table 1: Summary of Recent Comparative Studies (2024-2025)
| Pathogen | Phenotypic Method | Genotypic Method | Key Performance Findings | Study Reference |
|---|---|---|---|---|
| Acinetobacter baumannii (104 isolates) | MBL-E test, modified Hodge test | Detection of OXA-48, NDM, VIM genes | Genotypic detection: 60%; Phenotypic detection range: 36.54%-89.42%; Genotypic method noted as more time-effective. [12] | Bhavna S Pate et al. (2025) |
| Pasteurella multocida (80 isolates) | Disk diffusion, Broth microdilution | Whole-genome sequencing | Broth microdilution (MIC) showed a stronger positive correlation with genotypic results than disk diffusion. Correlation was strong for phenicols, tetracyclines, FQs but weak for sulfamethoxazole, β-lactams. [11] | Antibiotics (2025) |
| MDR Pseudomonas aeruginosa (183 isolates) | Broth microdilution (reference) | AMRFinderPlus | Categorical Agreement (CA) with BMD: Sensititre panel: 90.1-95.8%; Phoenix panel: 83.0-85.7%; Genotype: 74.9-91.9%. Genotypic prediction had high very major error rates due to uncharacterized mechanisms. [96] | Clayton W Hall et al. (2025) |
| Nocardia spp. (148 isolates) | Broth microdilution (MIC) | Whole-genome sequencing | Strong genotype-phenotype correlations for specific agents (e.g., sul1 & SXT, blaAST-1 & β-lactams). Also identified species-specific resistance genes and gyrA mutations linked to ciprofloxacin resistance. [97] | Front. Cell. Infect. Microbiol. (2025) |
Table 2: Analysis of Method Advantages and Limitations
| Feature | Phenotypic Testing | Genotypic Testing |
|---|---|---|
| Fundamental Principle | Measures observable bacterial growth inhibition in the presence of antimicrobials. [14] | Detects specific resistance genes (e.g., blaNDM, sul2) or mutations (e.g., in gyrA). [12] [11] |
| Turnaround Time | Conventional: 16-24 hours; Rapid systems (e.g., Selux DX): ~5.5 hours. [98] | Can be performed in a few hours, offering a significant time advantage over conventional phenotyping. [12] [95] |
| Key Advantage | Functional, comprehensive profile of resistance regardless of genetic mechanism; considered the gold standard. [95] [96] | Rapid results, high sensitivity/specificity for known mechanisms; detects resistance before expressible phenotype emerges. [12] [95] |
| Primary Limitation | Slower, as it requires bacterial growth. [14] | Cannot detect novel or uncharacterized resistance mechanisms; provides a prediction of resistance, not a direct measure. [96] [14] |
| Therapeutic Relevance | Directly measures the effect of antibiotics on the bacterium, which closely correlates with clinical outcome. | Predicts resistance based on the presence of known markers; clinical relevance depends on established genotype-phenotype correlations. [96] |
Experimental Protocols
To ensure reproducibility in comparative resistance studies, detailed protocols for reference phenotypic and core genotypic methods are essential. The following sections describe standardized workflows.
Protocol 1: Reference Broth Microdilution for Phenotypic AST
This protocol describes the reference broth microdilution method per CLSI standards, used for establishing minimum inhibitory concentration (MIC) against fastidious and infrequently isolated bacteria [10] [97].
3.1.1. Applications and Principle
- Application: Gold-standard phenotypic AST for a wide range of bacteria, including Nocardia spp., Pasteurella multocida, and other aerobes.
- Principle: A standardized bacterial inoculum is incubated in a microtiter plate containing serial two-fold dilutions of antibiotics. The MIC is the lowest concentration that completely inhibits visible growth after a specified incubation period.
3.1.2. Materials and Reagents
- Cation-adjusted Mueller-Hinton Broth (CAMHB) or other media specified by CLSI M07.
- Sterile, polystyrene, non-treated 96-well microtiter plates.
- Antibiotic stock solutions: Prepare from USP-grade powder or use commercial panels (e.g., Sensititre RAPMYCOI for Nocardia).
- Sterile saline (0.85% NaCl) or water.
- Adjustable pipettes and sterile tips.
- Incubator capable of maintaining 35±2°C in ambient air.
3.1.3. Step-by-Step Procedure
- Bacterial Inoculum Preparation:
- Pick 3-5 well-isolated colonies from an overnight (18-24 hour) culture plate.
- Suspend colonies in saline and vortex to achieve a homogeneous suspension.
- Adjust the turbidity to a 0.5 McFarland standard (approximately 1-2 x 10^8 CFU/mL for most organisms).
- Further dilute the suspension in broth to achieve the final working inoculum (typically 5 x 10^5 CFU/mL).
Plate Inoculation:
- Using a multichannel pipette, transfer 100 µL of the adjusted inoculum into each well of the antibiotic-containing microdilution panel. Include growth control (broth + inoculum) and sterility control (broth only) wells.
- Seal the plate with a adhesive cover to prevent evaporation.
Incubation:
- Incubate the plate for 16-20 hours at 35±2°C for most bacteria. Fastidious organisms like Nocardia may require 48-72 hours of incubation [97].
- Do not stack plates more than four high to ensure even heating.
Reading and Interpretation:
- Read the MIC visually or with a automated plate reader. The MIC is the lowest antibiotic concentration that completely inhibits visible growth.
- Compare results to CLSI M100 or other appropriate breakpoints for categorical interpretation (Susceptible, Intermediate, Resistant) [99].
Protocol 2: Whole-Genome Sequencing for Genotypic Resistance Profiling
This protocol outlines the process of using whole-genome sequencing (WGS) to identify antimicrobial resistance genes (ARGs) and mutations in bacterial isolates [11] [97].
3.2.1. Applications and Principle
- Application: Comprehensive, hypothesis-free detection of known ARGs and single-nucleotide polymorphisms (SNPs) associated with resistance.
- Principle: High-throughput sequencing of the entire bacterial genome, followed by bioinformatic alignment and comparison against curated resistance databases (e.g., CARD, BV-BRC).
3.2.2. Materials and Reagents
- DNA extraction kit (e.g., Wizard Genomic DNA Purification Kit, Promega).
- Quantification instrument (e.g., NanoDrop, Thermo Fisher Scientific).
- Illumina-compatible WGS library preparation kit.
- Illumina sequencing platform (e.g., NovaSeq) or equivalent.
- High-performance computing cluster with bioinformatics software.
3.2.3. Step-by-Step Procedure
- Genomic DNA Extraction:
- Harvest bacterial cells from a pure, fresh culture by centrifugation.
- Extract high-quality, high-molecular-weight genomic DNA using the selected kit, following the manufacturer's instructions.
- Assess DNA purity and concentration using a spectrophotometer. High-quality DNA should have an A260/A280 ratio of ~1.8-2.0.
Whole-Genome Sequencing:
- Prepare the sequencing library using the Illumina-compatible kit, fragmenting DNA and attaching platform-specific adapters.
- Sequence the library on the Illumina platform to achieve a minimum of 100x coverage (e.g., PE150 mode) [97].
- Perform initial quality control on raw reads using tools like Fastp v0.23.4 to trim adapters and remove low-quality bases.
Bioinformatic Analysis:
- De novo assembly of quality-filtered reads using an assembler like SPAdes v3.15.5.
- Annotate the assembled genome using Prokka v1.12.
- Identify ARGs and point mutations by comparing the genome to databases using the Resistance Gene Identifier (RGI) against the Comprehensive Antibiotic Resistance Database (CARD) or similar tools integrated into the BV-BRC platform [11] [97].
Workflow Visualization and Decision Pathways
The following diagrams illustrate the core workflows for comparative studies and the logical process for selecting the appropriate testing method.
Title: Comparative AST Study Workflow
Title: AST Method Selection Guide
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the protocols above requires specific, quality-assured reagents and platforms. The following table details key solutions for conducting these comparative studies.
Table 3: Research Reagent Solutions for AST Studies
| Item Name | Function/Application | Example Product/Reference |
|---|---|---|
| Sensititre Microdilution Panels | Pre-configured, dehydrated antibiotic panels for reference broth microdilution MIC testing. | Sensititre RAPMYCOI Panel (for Nocardia & aerobic Actinomycetes) [97]. |
| CAMHB Media | Culture medium for broth microdilution, adjusted for cation concentration to ensure accurate antibiotic activity. | Commercial CAMHB per CLSI standard M07 [10]. |
| DNA Extraction Kit | For purification of high-quality, inhibitor-free genomic DNA from bacterial cultures prior to WGS. | Wizard Genomic DNA Purification Kit (Promega) [97]. |
| CARD & BV-BRC | Curated bioinformatics databases for linking genetic determinants to antibiotic resistance phenotypes. | Comprehensive Antibiotic Resistance Database (CARD); Bacterial & Viral Bioinformatics Resource Center (BV-BRC) [11] [97]. |
| FDA/CDC AR Bank Isolates | Characterized reference strains with well-defined resistance mechanisms for assay validation/verification. | CDC & FDA Antibiotic Resistance Isolate Bank [10] [99]. |
| Selux Dx NGP System | Automated, rapid phenotypic AST system that significantly reduces time-to-result. | Selux Dx Next-Generation Phenotyping System [98]. |
| CLSI M100 Document | Standard for current, evidence-based interpretive breakpoints for AST. | CLSI M100 35th Edition (or latest) [99]. |
Within the field of antimicrobial and antiviral research, a fundamental challenge is accurately predicting how pathogens will respond to treatment. Two complementary methodologies have emerged to address this: genotypic testing, which identifies specific genetic mutations known as genetic determinants, and phenotypic testing, which directly measures a pathogen's ability to grow in the presence of a drug, resulting in an observable resistance profile. This Application Note provides a detailed framework for performing robust correlation analysis to bridge these two data types. Such integration is critical for validating the clinical significance of genetic mutations, improving interpretation systems, and guiding the development of effective treatment strategies, particularly within the context of HIV and antibiotic-resistant bacterial infections [100] [87].
Background: Genotypic and Phenotypic Resistance Testing
Resistance testing methodologies provide distinct but interrelated data. Understanding their core principles is a prerequisite for meaningful correlation analysis.
Genotypic Assays focus on identifying specific mutations within the pathogen's genome. For HIV, this involves sequencing the protease (PR), reverse transcriptase (RT), and integrase (IN) genes and comparing them to a reference wild-type strain to document mutations [100] [87]. The data output is typically a series of binary variables (e.g., presence or absence of mutations 41L, 65R, 67N, 184V) or a list of amino acid changes [100].
Phenotypic Assays directly measure the impact of these genetic changes on drug susceptibility. These tests quantify the concentration of a drug required to inhibit viral or bacterial replication by 50% (IC50) or 90% (IC90) in a controlled laboratory environment. The result is often expressed as a Fold Change (FC) in IC50 compared to a reference susceptible strain [100] [101]. Phenotypic testing provides a functional readout of the net effect of all mutations present in the genome.
Table 1: Core Characteristics of Resistance Testing Methodologies
| Feature | Genotypic Testing | Phenotypic Testing |
|---|---|---|
| What is Measured | Presence of specific nucleotide/amino acid mutations | Concentration of drug needed to inhibit pathogen replication (IC50/IC90) |
| Primary Output | List of mutations (e.g., K103N, M184V) | Fold-change in drug susceptibility relative to wild-type |
| Key Advantage | High sensitivity, fast turnaround, lower cost | Direct functional measure of resistance, integrates the effect of complex mutation patterns |
| Key Limitation | Requires prior knowledge to interpret mutation significance; may miss novel mutations | More complex, time-consuming, and costly; requires viable pathogen culture |
Computational and Statistical Framework for Correlation Analysis
Correlating genotypic and phenotypic data requires specialized statistical approaches to account for the nature of the data and avoid spurious findings.
Data Preprocessing and Management
A critical first step is ensuring consistency in genotypic data referencing. Different laboratories may use different reference strains (e.g., HXB2, NL4-3, Consensus B), which can lead to discrepancies in codon numbering. When merging datasets, it is crucial to use the exact nucleotide triplet and corresponding amino acid at each codon to avoid confusion [100]. Furthermore, clinical and laboratory metadata, such as detailed treatment history and timing of sample collection relative to therapy, are essential for contextualizing the resistance data and making informed assumptions during analysis [100] [87].
Statistical Modeling Approaches
Regression models are commonly employed to identify relationships between genetic mutations and the phenotypic fold-change. The phenotypic FC (a continuous variable) can be regressed against the genotypic mutations (binary predictor variables). Given the high dimensionality of genotypic data, with many potential mutations tested, the risk of false-positive associations (Type I error) is significant. It has been demonstrated that performing as few as 15 non-pre-selected statistical tests virtually guarantees a false-positive finding using a standard P-value threshold of 0.05 [100].
To address this, rigorous multiple testing corrections (e.g., Bonferroni, Benjamini-Hochberg) must be applied. Furthermore, findings from exploratory analyses should be treated as hypothesis-generating and require validation in independent datasets [100]. More advanced frameworks, such as those leveraging multi-type branching process models, can be used to deconvolute complex dynamics like therapy-induced resistance from bulk population data, providing a more robust inference of underlying mechanisms [102].
Application Notes & Experimental Protocols
Protocol 1: HIV-1 Drug Resistance Correlation Study
This protocol outlines the steps for correlating HIV-1 genotypic data with phenotypic resistance profiles.
I. Research Reagent Solutions
Table 2: Essential Reagents for HIV-1 Resistance Correlation Studies
| Reagent / Material | Function / Application |
|---|---|
| Patient Plasma Samples | Source of viral RNA for genotypic and phenotypic analysis. Must be processed or stored at -80°C. |
| Viral RNA Extraction Kit | Isolates high-quality HIV-1 RNA for subsequent molecular analysis. |
| RT-PCR & PCR Reagents | Amplifies the entire HIV-1 pol gene (PR, RT, IN) for sequencing and phenotypic testing. |
| Next-Generation Sequencing (NGS) Kit | Provides deep sequencing of the pol gene to detect low-frequency variants. |
| Phenotypic Assay Kit (e.g., PhenoSense) | Uses recombinant virus technology to measure drug susceptibility (IC50) against a panel of ARVs. |
| Cell Lines (e.g., 293T, TZM-bl) | Used for generating recombinant virus and quantifying viral replication in phenotypic assays. |
II. Step-by-Step Workflow
- Sample Collection & RNA Extraction: Collect plasma from patients with a viral load typically >500 copies/mL. Extract viral RNA using a commercial kit [87].
- Genotypic Analysis (Comprehensive pol Amplification):
- Amplify the entire pol region (PR, RT, IN) using a validated touchdown PCR protocol with primers designed to amplify a wide range of HIV-1 group M subtypes [103].
- Sequence the amplified product using Sanger sequencing or, for greater sensitivity, NGS to identify low-abundance variants.
- Analyze Sequences by aligning them to a consensus reference strain (e.g., HXB2) and catalog all amino acid differences from the wild-type.
- Phenotypic Analysis (Recombinant Virus Assay):
- Generate Recombinant Virus by inserting the patient-derived PR-RT-IN sequences into a HIV-1 vector backbone lacking these genes.
- Measure Drug Susceptibility by inoculating the recombinant virus into cell lines in the presence of a serial dilution of antiretroviral drugs.
- Calculate IC50/FC by determining the drug concentration that inhibits viral replication by 50% and comparing it to the IC50 of a reference wild-type virus [101].
- Data Integration & Correlation Analysis:
- Compile Data into a structured dataset pairing the list of mutations for each sample with its corresponding phenotypic FC values for each drug.
- Perform Statistical Analysis using multiple linear regression, with phenotypic FC as the dependent variable and mutations as independent variables. Apply multiple testing corrections.
- Validate Findings by comparing the correlation results with publicly available databases like the Stanford HIVdb and assess the clinical predictive value of identified mutations.
The following workflow diagram illustrates this integrated experimental process:
Protocol 2: Phenotypic Confirmatory Testing for Novel Mutations
When a novel genetic mutation is identified through statistical association, this protocol outlines the steps for functional validation.
I. Research Reagent Solutions
- Site-Directed Mutagenesis (SDM) Kit: Used to introduce the specific novel mutation into a wild-type molecular clone.
- HIV-1 Molecular Clone: A full-length infectious DNA clone of HIV-1 (e.g., pNL4-3).
- Tissue Culture Reagents & Antiretroviral Drugs: For cell culture maintenance and preparation of drug stocks for phenotypic testing.
II. Step-by-Step Workflow
- Generate Mutant Virus: Using an SDM kit, engineer the putative resistance mutation into a wild-type HIV-1 molecular clone. Verify the sequence.
- Produce Viral Stocks: Transfect the wild-type and mutant plasmid DNA into 293T cells to produce recombinant virus particles.
- Phenotypic Characterization: Titrate the virus and perform a phenotypic drug susceptibility assay as described in Protocol 1, comparing the IC50 of the mutant virus to the wild-type control.
- Replication Capacity Assessment: Measure the replication kinetics of the mutant virus in the absence of drug compared to the wild-type virus, as this can impact its clinical relevance [101].
The logical relationship and decision process in this confirmatory analysis is shown below:
Data Interpretation and Clinical Translation
The ultimate goal of correlation analysis is to improve patient care by accurately predicting treatment outcomes.
Building and Validating Interpretation Systems (IS): The correlations established through the above protocols feed into expert-defined interpretation systems (e.g., Stanford HIVdb, ANRS, Rega). These systems translate a list of mutations into a categorical prediction of "susceptible," "intermediate," or "resistant" for each drug [100]. It is important to note that different IS can yield discordant interpretations, underscoring the need for continuous refinement based on new clinical correlation data [100] [87].
Linking to Virological Outcomes: The most robust validation of a genotype-phenotype correlation is its ability to predict virological response in patients. Statistical analyses should evaluate how well the genotypic and/or phenotypic resistance measures predict the change in viral load at a defined time point (e.g., 8 or 24 weeks) after starting a new regimen [100]. This requires collaboration between statisticians, virologists, and clinicians to ensure that assumptions about the persistence of archived resistance and the selection of virological endpoints are sound [100].
The accurate detection of antimicrobial resistance (AMR) is a critical component of modern healthcare, directly impacting patient outcomes and public health initiatives. Research and clinical diagnostics increasingly rely on two complementary approaches: phenotypic testing, which measures the observable effects of an antimicrobial on a bacterial isolate, and genotypic testing, which identifies the genetic determinants of resistance. The validation of assays based on these methods must be conducted within robust regulatory frameworks to ensure their safety, efficacy, and reliability. For researchers and developers, understanding the regulatory pathways—primarily the U.S. Food and Drug Administration (FDA) and the European Union's CE-In Vitro Diagnostic (CE-IVD) marking under the In Vitro Diagnostic Regulation (IVDR)—is essential for successful technology translation and global market access. This document outlines the key regulatory requirements and provides detailed protocols for the validation of AMR assays, with a specific focus on the context of phenotypic versus genotypic resistance testing.
Comparative Regulatory Frameworks
Navigating the regulatory landscape is a fundamental step in the development of any diagnostic assay. The following section provides a high-level comparison of the two major regulatory systems.
Table 1: Comparison of Key FDA and EU IVDR Regulatory Features for AMR Assays
| Feature | U.S. FDA Pathway | EU CE-IVD (IVDR) Pathway |
|---|---|---|
| Governing Body | U.S. Food and Drug Administration (FDA) [104] | Notified Bodies (overseeing manufacturer compliance) [105] |
| Legal Basis | Federal Food, Drug, and Cosmetic Act [106] | Regulation (EU) 2017/746 (IVDR) [107] |
| Risk Classification | 3 classes (Class I, II, III) based on patient risk [104] | 4 classes (Class A, B, C, D) based on patient and public health risk [105] |
| Core Premarket Pathway | 510(k), De Novo, or Premarket Approval (PMA) [104] | Conformity assessment involving technical documentation review [105] |
| Clinical Evidence Focus | Analytical and clinical performance for intended use [104] | Performance Evaluation Report (clinical evidence and analytical performance) [107] |
| Post-Market Surveillance | Medical Device Reporting (MDR), complaint files [106] | Periodic Safety Update Report (PSUR) for Class C/D, Post-Market Surveillance Report (PMSR) for Class A/B [105] |
| Unique System | Unique Device Identification (UDI) [105] | European Database on Medical Devices (EUDAMED) [107] |
The U.S. FDA Pathway
In the United States, In Vitro Diagnostic (IVD) devices are regulated as medical devices by the FDA. The regulatory strategy is predicated on a risk-based classification system [104]:
- Class I: Low to moderate risk. Subject to general controls (e.g., establishment registration, device listing, quality system regulation).
- Class II: Moderate to high risk. Typically requires a premarket notification (510(k)) to demonstrate substantial equivalence to a legally marketed predicate device.
- Class III: High risk. Generally requires Premarket Approval (PMA), involving a rigorous scientific and regulatory review to demonstrate safety and effectiveness.
A significant recent development is the FDA's final rule on Laboratory Developed Tests (LDTs), issued in May 2024. This rule phases out the FDA's historical enforcement discretion for LDTs—tests designed, manufactured, and used within a single CLIA-certified laboratory—over a four-year period [106]. This means many laboratory-developed phenotypic AST or genotypic resistance tests will need to comply with FDA device regulations according to a defined timeline [106]:
- Stage 1 (By May 2025): Compliance with medical device reporting and quality system complaint files.
- Stage 5 (By May 2028): Compliance with premarket review requirements for moderate and low-risk IVDs.
For public health emergencies, the FDA has issued draft guidance outlining validation recommendations for IVDs during a declared emergency under section 564 of the FD&C Act [108].
The EU CE-IVD Pathway under IVDR
The European Union's In Vitro Diagnostic Regulation (IVDR 2017/746) represents a significant overhaul of the previous regulatory framework, with full application coming into force in 2025-2027 [107]. The IVDR introduces a stronger emphasis on life-cycle management of devices, clinical evidence, and post-market surveillance. Key aspects include:
- Risk-Based Classification: IVDs are classified from Class A (lowest risk) to Class D (highest risk) based on rules defined in Annex VIII of the IVDR. AMR assays often fall into higher-risk classes (C or D) due to their impact on critical treatment decisions [105].
- Notified Body Oversight: Approximately 80% of IVDs, including most AMR tests, now require confirmation of conformity by a Notified Body, a stark increase from about 20% under the previous directive [107].
- Performance Evaluation: Under IVDR, manufacturers must establish and maintain a Performance Evaluation Report that continuously justifies conformity based on three pillars: Analytical Performance, Clinical Performance, and Scientific Validity [105]. This is a continuous process throughout the device's lifecycle.
Table 2: Key Performance Evaluation Components under EU IVDR for AMR Assays
| Performance Pillar | Definition | Examples for Phenotypic AST | Examples for Genotypic AMR |
|---|---|---|---|
| Scientific Validity | The association of an analyte with a clinical condition or physiology | Link between MIC and clinical resistance [5] | Link between a specific AMR gene (e.g., mecA) and resistance to an antimicrobial (e.g., methicillin) [109] |
| Analytical Performance | The ability of a device to correctly detect or measure an analyte | Precision, accuracy (trueness), linearity, limit of detection for MIC panels [110] | Sensitivity, specificity, reproducibility, limit of detection for nucleic acid amplification or sequencing assays [109] |
| Clinical Performance | The ability of a device to yield results correlating with a clinical condition | Concordance of categorical result (S/I/R) with patient treatment outcome | Concordance of detected resistance gene with phenotypic resistance outcome in a target population [110] |
Quality Standards and Validation Frameworks
A robust Quality Management System (QMS) is the foundation for regulatory compliance. The international standard ISO 13485:2016 specifies requirements for a QMS for medical device manufacturers and is harmonized with both FDA requirements and the EU IVDR [107]. Key elements include management responsibility, resource management, product realization, and measurement, analysis, and improvement.
The FDA's Quality System Regulation (21 CFR Part 820) is transitioning to align with ISO 13485:2016 through the Quality Management System Regulation (QMSR) to reduce duplication for manufacturers operating globally [111]. Key principles for assay validation under these frameworks include:
- Risk-Based Approach: Validation activities should be proportionate to the risk associated with the device and its intended use.
- Lifecycle Approach: Validation is not a one-time event but continues throughout the product lifecycle via activities like Continued Process Verification (CPV) and Post-Market Surveillance (PMS) [112].
- Data Integrity: All data generated during validation must be accurate, complete, and traceable, adhering to principles of ALCOA+ (Attributable, Legible, Contemporaneous, Original, and Accurate) [112].
Experimental Protocols for AMR Assay Validation
The following protocols provide detailed methodologies for validating key aspects of both phenotypic and genotypic AMR tests.
Protocol 1: Broth Microdilution for Phenotypic Susceptibility Testing
This protocol describes the reference method for determining the Minimum Inhibitory Concentration (MIC) of antimicrobials against bacterial isolates, establishing the phenotypic resistance profile [110].
1. Principle The MIC is the lowest concentration of an antimicrobial that prevents visible growth of a microorganism. A standardized inoculum is incubated with serial dilutions of an antimicrobial in a broth medium. Visual or automated reading after a defined incubation period determines the MIC, which is interpreted using established clinical breakpoints (e.g., from EUCAST or CLSI).
2. Research Reagent Solutions Table 3: Essential Reagents for Broth Microdilution
| Item | Function |
|---|---|
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized growth medium ensuring consistent cation concentrations that affect antimicrobial activity. |
| Sterile 96-Well Microdilution Trays | Pre-prepared trays with serial two-fold dilutions of antimicrobials or empty trays for manual preparation. |
| Turbidity Standard (0.5 McFarland) | Reference for standardizing the density of the bacterial inoculum suspension. |
| Sterile Saline or Water | Diluent for adjusting bacterial inoculum density. |
| Quality Control Strains | Reference strains with known MIC ranges (e.g., E. coli ATCC 25922, S. aureus ATCC 29213) to verify test performance. |
3. Procedure
- Step 1: Inoculum Preparation. Pick 3-5 well-isolated colonies from an overnight agar plate. Suspend in saline and adjust the turbidity to a 0.5 McFarland standard (~1-2 x 10^8 CFU/mL). Further dilute the suspension in broth to achieve the final testing inoculum of ~5 x 10^5 CFU/mL.
- Step 2: Inoculation of Panel. Within 15-30 minutes of adjusting the inoculum, add a precise volume (e.g., 100 µL) of the standardized inoculum to each well of the microdilution tray containing the antimicrobial dilutions. Include growth control (inoculum, no drug) and sterility control (broth only) wells.
- Step 3: Incubation. Seal the tray and incubate under appropriate conditions (typically 35±2°C in ambient air for 16-20 hours).
- Step 4: Reading and Interpretation. Examine the wells for visible growth. The MIC is the lowest antimicrobial concentration that completely inhibits growth. Compare the MICs of quality control strains to their accepted ranges to validate the run.
4. Validation Parameters
- Precision: Determine intra-assay (repeatability) and inter-assay (reproducibility) variability by testing a panel of isolates multiple times.
- Accuracy: Compare MIC results to a reference method or expected results from quality control strains.
- Categorical Agreement: Assess the agreement of interpretive categories (Susceptible, Intermediate, Resistant) with a reference method.
Protocol 2: Whole-Genome Sequencing for Genotypic Resistance Prediction
This protocol outlines the use of Whole-Genome Sequencing (WGS) to identify known antimicrobial resistance genes (ARGs) and mutations, predicting the genotypic resistance profile [109].
1. Principle Genomic DNA is extracted from a bacterial isolate and sequenced using a high-throughput platform (e.g., Illumina). The resulting sequencing reads are assembled, and the genome is analyzed against curated AMR gene databases (e.g., ResFinder, CARD) to identify the presence of genetic determinants conferring resistance.
2. Research Reagent Solutions Table 4: Essential Reagents for WGS-Based AMR Detection
| Item | Function |
|---|---|
| DNA Extraction Kit | For high-quality, high-purity genomic DNA isolation from bacterial cultures. |
| DNA Quantitation Kit (Fluorometric) | For accurate quantification of DNA concentration prior to library preparation. |
| Library Preparation Kit | Prepares fragmented and adapter-ligated DNA for sequencing. |
| Sequencing Reagents & Flow Cell | Chemistry and solid support for the sequencing-by-synthesis reaction. |
| Bioinformatics Software/Platform | For quality control, genome assembly, and AMR gene/mutation detection (e.g., GalaxyTrakr, CLC Genomics). |
3. Procedure
- Step 1: DNA Extraction. Extract genomic DNA from a pure bacterial culture using a validated method. Ensure DNA integrity and purity (e.g., via spectrophotometry and gel electrophoresis).
- Step 2: Library Preparation and Sequencing. Quantify the DNA. Prepare a sequencing library according to the platform manufacturer's instructions (fragmentation, end-repair, adapter ligation, and amplification). Load the library onto the sequencer (e.g., Illumina MiSeq/NextSeq) and run.
- Step 3: Bioinformatic Analysis.
- Quality Control: Assess raw sequencing reads for quality (e.g., using FastQC) and trim adapters/low-quality bases.
- Genome Assembly: Assemble the trimmed reads into contigs using a de novo assembler (e.g., SPAdes).
- AMR Gene Identification: Use a tool like ResFinder or the AMRFinderPlus to compare the assembled genome against its database. A minimum identity threshold and coverage are set to call a positive hit.
- Step 4: Interpretation. Generate a report of detected AMR genes and mutations. Correlate these findings with expected phenotypic resistance profiles based on published literature.
4. Validation Parameters
- Sensitivity and Specificity of Prediction: Calculate the percentage of phenotypically resistant isolates correctly predicted by WGS (sensitivity) and the percentage of phenotypically susceptible isolates correctly predicted (specificity) [109] [110].
- Concordance: Determine the overall agreement between genotypic prediction and phenotypic AST results across a large set of isolates and antimicrobials [110].
- Limit of Detection: For direct-from-specimen tests, establish the lowest microbial load that can be reliably detected.
Workflow Diagram: Phenotypic vs. Genotypic AMR Testing Pathways
Diagram 1: A comparison of the core workflows for phenotypic and genotypic antimicrobial resistance testing.
Correlation of Phenotypic and Genotypic Data
A critical step in validating genotypic AMR assays is establishing a correlation with the phenotypic reference method. Studies show a high overall concordance (>90%) between WGS predictions and phenotypic AST for certain bacterial groups like Salmonella and Enterobacterales [109]. However, discordances occur and are informative.
Table 5: Analysis of Phenotype-Genotype Discordance in AMR Testing (based on [110])
| Discordance Type | Description | Potential Causes |
|---|---|---|
| Phenotypically Resistant, Genotypically Susceptible (R-S) | Phenotype shows resistance, but no known resistance gene/mutation is detected. | Novel or undetected mechanisms (e.g., new efflux pumps, enzyme variants); non-genetic factors (e.g., biofilm formation, permeability); limitations in database completeness [109]. |
| Phenotypically Susceptible, Genotypically Resistant (S-R) | A known resistance gene is present, but the isolate is phenotypically susceptible. | Silent or unexpressed genes; gene expression level is too low to confer resistance; fitness costs leading to loss of gene in vitro; specific genetic context required for expression (e.g., promoter strength) [5]. |
Successfully navigating the regulatory pathways for FDA and CE-IVD marking requires a strategic and proactive approach. For assays characterizing phenotypic and genotypic AMR, developers must integrate regulatory planning early in the R&D phase. Key to success is a deep understanding of the distinct but converging requirements: a risk-based classification, robust analytical and clinical performance studies that acknowledge the strengths and limitations of each method, a lifecycle approach to quality management, and a comprehensive post-market surveillance plan. As regulatory landscapes evolve—with the FDA's increased oversight of LDTs and the full implementation of the EU IVDR—a commitment to generating rigorous scientific validation data and maintaining detailed documentation remains the universal constant for ensuring patient safety and achieving global market access.
The escalating global threat of antimicrobial resistance (AMR) necessitates advanced diagnostic strategies that can deliver rapid, accurate, and clinically actionable results. Conventional antimicrobial susceptibility testing (AST) often relies on phenotypic methods, which, while considered the gold standard, can require a minimum of 72 hours from specimen collection to final results, impeding timely therapeutic decisions [14]. Genotypic methods offer speed but can lack comprehensive predictive power, as they may fail to detect diverse or novel resistance mechanisms; for instance, a carbapenemase gene is identifiable in fewer than 50% of bacteria found to be phenotypically carbapenem-resistant [14]. The integration of phenotypic and genotypic data represents a frontier in diagnostic microbiology, promising to overcome the limitations of either approach used in isolation. This paradigm is crucial for understanding intrinsic resistance, enhancing prediction accuracy, and ultimately improving patient outcomes. Framed within the broader context of phenotypic versus genotypic intrinsic resistance testing research, this article details practical protocols and strategies for their synergistic integration.
Background and Quantitative Landscape of AMR
The challenge of AMR extends beyond human medicine into the environment and wildlife. Non-human primates (NHPs), as close relatives of humans, serve as sentinels for zoonotic transmission and are potential natural reservoirs of resistant bacteria. A recent analysis of 37 studies revealed a concerning prevalence of antimicrobial-resistant bacteria (ARB) and antibiotic resistance genes (ARGs) in these species [113].
Table 1: Prevalence of Key Resistant Bacteria and Antibiotic Resistance in Non-Human Primates [113]
| Category | Specific Example | Prevalence / Notes |
|---|---|---|
| Common Resistant Bacteria | Staphylococcus spp. | 45.95% (across studied NHPs) |
| Escherichia spp. | 29.73% (across studied NHPs) | |
| Antibiotics with High Resistance | Tetracycline | 40.54% |
| Ciprofloxacin | 32.43% | |
| Erythromycin | 24.34% | |
| Common Resistance Genes (ARGs) | ermC, tetA, tetM | Widely distributed |
| aadA, aph(3″)-II, qnrS1 | Widely distributed |
This data underscores the complex ecology of AMR and highlights the need for sophisticated monitoring tools that can track its emergence and spread across different reservoirs, informing the One Health approach to resistance management [113].
Experimental Protocols for Data Integration
Protocol: Next-Generation Rapid Phenotypic AST
This protocol outlines the procedure for using novel technologies to significantly reduce the time-to-result for phenotypic AST, creating a faster baseline against which genotypic predictions can be validated and integrated [14].
I. Principle Conventional phenotypic AST requires isolating pure bacterial colonies from a clinical specimen, followed by incubation in the presence of antimicrobials, a process taking 4-24 hours after isolation. Next-generation rapid phenotypic AST technologies leverage innovations such as microfluidics, microscopy, and cytometry to detect subtle changes in bacterial growth or viability in the presence of antimicrobials in a fraction of the time, often within hours directly from positive blood cultures or other clinical samples [14].
II. Specimen Requirements
- Specimen Types: Positive blood culture bottles, urine, pure bacterial colonies.
- Specimen Processing: For positive blood cultures, a brief sub-culture or direct dilution may be required depending on the platform.
III. Reagent and Material Solutions
Table 2: Research Reagent Solutions for Rapid Phenotypic AST
| Item Name | Function/Description |
|---|---|
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized growth medium for AST, ensures consistent ion concentration for antibiotic activity. |
| Antimicrobial Panels | Pre-configured, serial dilutions of antibiotics in a 96-well plate format. |
| Viability Stains (e.g., SYTOX Green, Propidium Iodide) | Fluorescent dyes that penetrate compromised membranes of dead cells, enabling rapid viability counting. |
| Microfluidic Growth Chips | Cartridges with micro-channels that confine single or few bacteria, allowing for rapid growth monitoring via microscopy. |
IV. Step-by-Step Procedure
- Specimen Inoculation: Dilute the positive blood culture broth or suspend pure colonies in CAMHB to a standardized turbidity (e.g., 0.5 McFarland). Load the suspension into the dedicated cartridge or microfluidic device of the rapid AST system.
- Antimicrobial Exposure: The automated system introduces the bacterial suspension to pre-determined concentrations of antimicrobials.
- Incubation and Monitoring: Place the cartridge in the analyzer. The system incubates the sample and monitors bacterial growth or death in real-time using methods such as:
- Time-lapse Microscopy: Tracking micro-colony formation in microfluidic channels.
- Flow Cytometry: Analyzing single cells for viability stains.
- Morphological Analysis: Detecting changes in cell shape and size.
- Data Analysis and MIC Determination: The system's software analyzes growth kinetics over 2-5 hours. The Minimum Inhibitory Concentration (MIC) is determined by identifying the lowest antibiotic concentration that significantly inhibits growth compared to a negative control.
- Result Interpretation: The MIC is interpreted as Susceptible, Intermediate, or Resistant based on Clinical and Laboratory Standards Institute (CLSI) or EUCAST breakpoints.
V. Quality Control
- Run control strains (e.g., E. coli ATCC 25922, S. aureus ATCC 29213) with each batch.
- Ensure reagents meet expiration dates and storage conditions.
Protocol: Genotypic AST and Machine Learning Integration
This protocol describes the workflow for generating genotypic data and integrating it with phenotypic results using machine learning to build predictive models.
I. Principle This method involves extracting bacterial DNA, screening for known ARGs via PCR or whole-genome sequencing (WGS), and using the resulting genotypic profile alongside phenotypic AST data to train predictive machine learning models. The goal is to create a system that can accurately predict phenotypic resistance from genotype alone, even for complex resistance patterns.
II. Specimen Requirements
- Specimen Types: Pure bacterial biomass from culture.
- Specimen Processing: Centrifuge bacterial suspension to form a pellet for DNA extraction.
III. Reagent and Material Solutions
Table 3: Research Reagent Solutions for Genotypic AST and ML
| Item Name | Function/Description |
|---|---|
| Genomic DNA Extraction Kit | For high-quality, high-purity DNA extraction from bacterial pellets. |
| PCR Master Mix & Primers | For amplification of specific, pre-identified antibiotic resistance genes (e.g., mecA, blaKPC, ermC). |
| Whole-Metagenome Sequencing Reagents | For hypothesis-free sequencing of all genetic material in a sample, allowing discovery of novel ARGs. |
| Polymerase Chain Reaction (PCR) Thermocycler | Instrument for amplifying target DNA sequences. |
| Next-Generation Sequencing (NGS) Platform | For performing whole-genome sequencing. |
IV. Step-by-Step Procedure
- DNA Extraction: Use a commercial DNA extraction kit to isolate genomic DNA from the bacterial pellet. Quantify and assess DNA purity using a spectrophotometer.
- Genotypic Profiling:
- Option A (Targeted PCR): Perform multiplex PCR using panels of primers specific to common ARGs (e.g., tetA, ermC). Analyze amplicons via gel electrophoresis.
- Option B (WGS): Prepare a sequencing library from the extracted DNA and perform whole-genome sequencing on an NGS platform. Use bioinformatics pipelines to align sequences and identify ARGs in curated databases (e.g., CARD, ResFinder).
- Data Integration and Model Training:
- Compile a dataset where each bacterial isolate is represented by its genotypic profile (presence/absence of ARGs) and its phenotypic MIC profile.
- Employ machine learning algorithms (e.g., Gradient Boosting models like XGBoost or LightGBM) to train a classifier. The genotype data serves as the input features, and the phenotypic susceptibility result (S/I/R) is the target variable [114].
- To mitigate bias from imbalanced datasets (e.g., over-representation of certain resistance types), use techniques like population-conditional re-sampling [114].
- Model Validation: Validate the trained model's performance on a hold-out test set of isolates not used in training. Report metrics including accuracy, categorical agreement, and major error rates.
V. Quality Control
- Include positive and negative controls in all PCR runs.
- Use standardized bioinformatics pipelines and reference databases for WGS analysis.
Visualization of Integrated Workflows
The following diagrams, created using the specified color palette, illustrate the core logical relationships and experimental workflows described in these protocols.
Diagram 1: Data Integration and Modeling Workflow
Diagram 2: Combined Clinical Testing Pathway
Conclusion
Phenotypic and genotypic resistance testing are complementary pillars in the fight against antimicrobial resistance. While phenotypic methods provide a direct measure of microbial behavior under therapeutic pressure, genotypic approaches offer rapid detection of resistance mechanisms and insights into evolutionary pathways. The future lies in integrated solutions that combine the functional validation of phenotyping with the speed and depth of genotypic analysis, accelerated by advancements in sequencing, microfluidics, and data analytics. For researchers and drug developers, this synergy is crucial for advancing diagnostic precision, guiding antimicrobial stewardship, and developing next-generation therapeutics to address the escalating global AMR crisis.
References
- 1. Phenotype vs Genotype: Understanding Genetic ... [https://www.cd-genomics.com/blog/phenotype-vs-genotype-understanding-genetic-expression-and-its-implications/]
- 2. Genotype [https://en.wikipedia.org/wiki/Genotype]
- 3. Phenotype [https://en.wikipedia.org/wiki/Phenotype]
- 4. Genotype–phenotype distinction [https://en.wikipedia.org/wiki/Genotype%E2%80%93phenotype_distinction]
- 5. What Is The Difference Between Phenotypic Versus ... [https://www.idstewardship.com/difference-phenotypic-versus-genotypic-resistance/]
- 6. Genotype and Phenotype Interactions: Foundations, ... [https://www.cd-genomics.com/microbioseq/resource-genotype-phenotype-interactions-applications.html]
- 7. Phenotypic Plasticity: From Theory and Genetics to Current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7198268/]
- 8. Genotyping vs. Phenotyping | Monogram Biosciences - Labcorp [https://monogrambio.labcorp.com/resources/genotyping-vs-phenotyping]
- 9. Advancements in AI-driven drug sensitivity testing research [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1560569/full]
- 10. Major updates to FDA-recognized Clinical and Laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077184/]
- 11. Comparative Analysis of Phenotypic and Genotypic ... [https://www.mdpi.com/2079-6382/14/9/906]
- 12. Comparison of Phenotypic and Genotypic Detection ... [https://pubmed.ncbi.nlm.nih.gov/40027060/]
- 13. Clinical impact of rapid test for macrolide-resistance gene ... [https://pubmed.ncbi.nlm.nih.gov/40829438/]
- 14. Next-generation rapid phenotypic antimicrobial ... [https://www.nature.com/articles/s41467-024-53930-x]
- 15. Impeding pathways of intrinsic resistance in Escherichia coli ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12543169/]
- 16. Clinical impact of rapid test for macrolide-resistance gene ... [https://www.sciencedirect.com/science/article/pii/S1876034125002795]
- 17. A Six-Step Protocol for Monitoring Antimicrobial Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566455/]
- 18. Overview of Quantitative Methodologies to Understand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7459578/]
- 19. Development of a Protocol for Predicting Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375328/]
- 20. Evolution of to Irinotecan in Cancer Cells Involves... Resistance [https://pmc.ncbi.nlm.nih.gov/articles/PMC10218574/]
- 21. GWAS and functional studies suggest a role for altered DNA repair in... [https://elifesciences.org/articles/75860]
- 22. Phenotypic and genotypic characterization of antibiotic- ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1649307/full]
- 23. Genotypic and Phenotypic Methods in the Detection of MDR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12292002/]
- 24. Impeding pathways of intrinsic resistance in Escherichia coli ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003443]
- 25. Specimen Collection Practices for Microbiologic Culture [https://health.ucdavis.edu/blog/lab-best-practice/specimen-collection-practices-for-microbiologic-culture/2020/07]
- 26. Advancing Drug Resistance Detection: Comparative ... [https://www.mdpi.com/2813-9038/2/3/14]
- 27. How AI can help us beat AMR | npj Antimicrobials and ... [https://www.nature.com/articles/s44259-025-00085-4]
- 28. Global antibiotic resistance surveillance report 2025 [https://www.who.int/publications/i/item/9789240116337]
- 29. Project funded to accelerate testing for respiratory infections [https://www.imperial.ac.uk/news/articles/medicine/infectious-disease/2025/project-funded-to-accelerate-testing-for-respiratory-infections/]
- 30. Joint transnational call 2026 [https://ohamr.eu/calls/call-2026-new-treatments-to-tackle-amr/]
- 31. Antimicrobial Susceptibility Testing: A Comprehensive Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9024665/]
- 32. The Genotype-to-Phenotype Dilemma: How Should ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8316082/]
- 33. Performance of disc diffusion and gradient strip test on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12494204/]
- 34. Broth Microdilution and Gradient Diffusion Strips vs. ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388896/]
- 35. Implementation and Evaluation of Gradient Strip ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10210089/]
- 36. Clinical Breakpoint Tables [https://www.eucast.org/bacteria/clinical-breakpoints-and-interpretation/clinical-breakpoint-tables/]
- 37. Notices of Updates [https://www.fda.gov/drugs/development-resources/notices-updates]
- 38. Prediction of live birth - selection of embryos using morphokinetic ... [https://pubmed.ncbi.nlm.nih.gov/36622075/]
- 39. Assessment of artificial intelligence model and manual morphokinetic ... [https://rbej.biomedcentral.com/articles/10.1186/s12958-024-01198-7]
- 40. 微流控专题| 单细胞封装技术全解析:从原理到应用的前沿探索 [https://dichbio.com/single-cell-encapsulation-technology-in-its-entirety-a-cutting-edge-exploration-from-principle-to-application/]
- 41. 基于光学检测的分子间相互作用表征技术研究进展 [https://html.rhhz.net/dejydxxb/html/2024/7/20240712.htm]
- 42. Biba et al. A Comparison of Sanger Sequencing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12390554/]
- 43. How to Choose Suitable SNP Genotyping Method [https://www.cd-genomics.com/how-to-chose-suitable-snp-genotyping-method.html]
- 44. PCR, Sequencing or Array Genotyping: Select the Best ... [https://www.thermofisher.com/blog/behindthebench/pcr-sequencing-or-array-genotyping-select-the-best-dna-technology-platform-for-your-consumer-genetics-product/]
- 45. DNA Genotyping: How It Differs from Sequencing and ... [https://www.fjc.gov/content/361256/dna-genotyping-how-it-differs-sequencing-and-relevant-methods]
- 46. A simple and cost-effective SNP genotyping assay for ... [https://www.sciencedirect.com/science/article/pii/S0926669025002390]
- 47. DNA Microarray Market Size to Hit USD 6.13 Billion By 2034 [https://www.precedenceresearch.com/dna-microarray-market]
- 48. A Precision Approach to HCMV Drug Resistance [https://pubmed.ncbi.nlm.nih.gov/40580282/]
- 49. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 50. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 51. Short-Read Sequencing vs. Long ... [https://seqwell.com/short-read-sequencing-vs-long-read-sequencing-which-technology-is-right-for-your-research/]
- 52. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 53. Next-Generation Sequencing Illumina Workflow–4 Key Steps [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/illumina-workflow.html]
- 54. Sample Preparation for NGS – A Comprehensive Guide [https://frontlinegenomics.com/sample-preparation-for-ngs-a-comprehensive-guide/]
- 55. Joint processing of long- and short-read sequencing data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12296420/]
- 56. Making AST fast [https://myadlm.org/cln/articles/2025/mayjune/making-act-fast]
- 57. Emerging technologies for rapid phenotypic antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607566/]
- 58. head-to-head comparison of three rapid AST systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607571/]
- 59. head-to-head comparison of three rapid AST systems for ... [https://pubmed.ncbi.nlm.nih.gov/41065398/]
- 60. Rapid antimicrobial susceptibility testing (AST) based on in ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525017885]
- 61. An overview of the antimicrobial resistance mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6604941/]
- 62. Comparison of Phenotypic and Genotypic Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11871954/]
- 63. Phenotypic and Genotypic Profiling of Antimicrobial ... [https://www.sciencedirect.com/science/article/pii/S1542012425001247]
- 64. Intrinsic antibiotic resistance: Mechanisms, origins ... [https://www.sciencedirect.com/science/article/abs/pii/S1438422113000246]
- 65. Discovery - Automated Workflows Compound Screening in Drug [https://lifesciences.danaher.com/us/en/solutions/small-molecule-drug-discovery/preliminary-screening.html]
- 66. Assay Development : Best Practices in ... | Technology Networks Drug [https://www.technologynetworks.com/drug-discovery/articles/assay-development-329953]
- 67. 从头设计全新抗生素,精准杀灭耐药菌- 组学专区 [https://news.bioon.com/article/526d891956cf.html]
- 68. Making Drug Discovery More People-Centric: Developing Organoids... [https://www.linkedin.com/pulse/making-drug-discovery-more-people-centric-developing/]
- 69. Genotypic resistance determined by whole genome ... [https://www.nature.com/articles/s41598-023-27723-z]
- 70. A simple and cost-saving phenotypic drug susceptibility ... [https://www.nature.com/articles/srep33559]
- 71. Evaluation of Marker Gene-Based In Silico Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561268/]
- 72. Minimal gene signatures enable high-accuracy prediction ... [https://www.nature.com/articles/s41540-025-00584-0]
- 73. Comparison of genotypic and phenotypic antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10644917/]
- 74. A simple, low-cost, and highly efficient protocol for rapid ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1637776/full]
- 75. Sample preparation strategies for mass spectrometry ... [https://www.sciencedirect.com/science/article/pii/S0165993623002388]
- 76. Optimization of MALDI Matrices and Their Preparation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12059521/]
- 77. Antimicrobial Resistance Diagnostics Market Size, 2032 [https://www.persistencemarketresearch.com/market-research/antimicrobial-resistance-diagnostics-market.asp]
- 78. Antimicrobial Resistance Diagnostics Market Size, Share 2035 [https://www.transparencymarketresearch.com/antimicrobial-resistance-diagnostics-market.html]
- 79. Antibiotic Susceptibility Testing Devices Market [https://www.futuremarketinsights.com/reports/antibiotic-susceptibility-testing-devices-market]
- 80. Antimicrobial Resistance [https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/antimicrobial-resistance]
- 81. Bioinformatics approaches to the study of antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8683011/]
- 82. Comparative assessment of annotation tools reveals ... [https://www.nature.com/articles/s41598-025-24333-9]
- 83. Genotypic and phenotypic comparison of drug resistance profiles of clinical multidrug-resistant Mycobacterium tuberculosis isolates using whole genome sequencing in Latvia | BMC Infectious Diseases | Full Text [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-023-08629-7]
- 84. Pipeline validation for the identification of antimicrobial ... [https://www.nature.com/articles/s41598-023-42154-6]
- 85. Phenotypic Drug Discovery: Recent successes, lessons ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9708951/]
- 86. Opportunities and challenges in phenotypic drug discovery [https://www.nature.com/articles/nrd.2017.111]
- 87. HIV Resistance Assays - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK558864/]
- 88. HIV Resistance Assays - Clinical Guidelines ... [https://www.hivguidelines.org/guideline/hiv-resistance-assays/]
- 89. Role of phenotypic testing in determining the mechanism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6563753/]
- 90. 2.3. Phenotypic Antimicrobial Resistance Testing [https://bio-protocol.org/exchange/minidetail?id=16543759&type=30]
- 91. Rapid Phenotypic and Genotypic Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11351821/]
- 92. Molecular diagnostics for genotypic detection of antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9937039/]
- 93. CRE对多粘菌素B的敏感性及两种检测方法的差异 [http://zggrkzzz.ijournals.cn/html/zggrkzzz/2019/11/2019-11-1059.htm]
- 94. Establishment and Evaluation of a Resazurin-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10839469/]
- 95. Advances in genotypic antimicrobialresistance testing [https://pubmed.ncbi.nlm.nih.gov/39300049/]
- 96. Evaluation of phenotypic and genotypic susceptibility ... [https://pubmed.ncbi.nlm.nih.gov/41248977/]
- 97. Genomic and phenotypic characterization of antimicrobial ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1672889/full]
- 98. Evaluation of a Novel Rapid Phenotypic Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562109/]
- 99. Breakpoint Implementation Toolkit (BIT) | Resources [https://clsi.org/resources/breakpoint-implementation-toolkit/]
- 100. Understanding the statistical analysis of resistance data - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK2247/]
- 101. HIV phenotypic testing - Monogram Biosciences [https://monogrambio.labcorp.com/resources/phenotyping]
- 102. A statistical framework for detecting therapy-induced ... [https://www.nature.com/articles/s41540-025-00560-8]
- 103. Development of a new comprehensive HIV-1 genotypic ... [https://www.sciencedirect.com/science/article/pii/S1567134820300757]
- 104. In Vitro Diagnostic (IVD) Devices Complete Guide ... [https://www.greenlight.guru/blog/ivd-devices]
- 105. IVDs – A Comparison of Requirements between the US and EU [https://blog.pqegroup.com/medical-device/ivds-a-comparison-of-requirements-between-the-us-and-eu]
- 106. FDA Intends To Regulate Many Clinical Labs as Medical ... [https://www.arnoldporter.com/en/perspectives/advisories/2024/05/fdas-final-laboratory-developed-test-rule]
- 107. Navigating the IVD Regulatory Landscape in 2025 - Bioexcel [https://bioexcelife.com/navigating-the-ivd-regulatory-landscape-in-2025/]
- 108. Validation of Certain In Vitro Diagnostic Devices for ... [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/validation-certain-in-vitro-diagnostic-devices-emerging-pathogens-during-section-564-declared-emergency]
- 109. Comparison of genotypic and phenotypic antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11185115/]
- 110. One Day in Denmark: Comparison of Phenotypic and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.804627/full]
- 111. CE Mark Medical Device Regulatory Changes 2025 [https://mavenprofserv.com/blog/regulatory-changes-medical-device-manufacturers/]
- 112. The Future of US FDA Pharmaceutical Validation Services ... [https://confiancapharmazon.com/future-us-fda-pharmaceutical-validation-services-2025/]
- 113. Phenotypic and Genotype Patterns of Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561749/]
- 114. Machine Learning Strategies for Improved Phenotype ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10799683/]