Forward vs Reverse Chemogenomics: A Strategic Guide for Modern Drug Discovery
This article provides a comprehensive analysis of forward and reverse chemogenomics, two pivotal strategies reshaping target identification and validation.
Forward vs Reverse Chemogenomics: A Strategic Guide for Modern Drug Discovery
Abstract
This article provides a comprehensive analysis of forward and reverse chemogenomics, two pivotal strategies reshaping target identification and validation. Tailored for researchers and drug development professionals, it explores the foundational principles, distinct methodologies, and practical applications of each approach. The content delves into common challenges and optimization techniques, supported by real-world case studies. A direct comparative analysis equips scientists to select the appropriate strategy based on project goals, from probing unknown disease mechanisms to rationally designing modulators for specific protein families. This guide synthesizes traditional knowledge with cutting-edge advancements, including the role of AI and open-source initiatives like Target 2035, offering a roadmap for integrating these powerful techniques into next-generation therapeutic development.
Decoding the Core Principles: From Phenotype to Target and Back
Chemogenomics represents a systematic, genome-scale approach to drug discovery that integrates the screening of chemical libraries with the functional study of target families. The core premise of chemogenomics is the comprehensive exploration of chemical space against biological target space to identify novel therapeutics and simultaneously elucidate the function of previously uncharacterized targets [1]. This paradigm operates on the principle that focused chemical libraries containing known ligands for specific target families (e.g., GPCRs, kinases, proteases) are likely to contain compounds that interact with other members of the same family, enabling rapid identification of modulators for orphan targets [1]. The field has gained significant momentum with advancements in high-throughput screening technologies, functional genomics, and computational biology, allowing researchers to map the complex interactions between small molecules and biological systems at an unprecedented scale.
The completion of the human genome project provided an abundance of potential targets for therapeutic intervention, and chemogenomics strives to study the intersection of all possible drugs on all these potential targets [1]. This systematic approach represents a significant shift from traditional one-target-one-drug discovery methods toward a more integrated strategy that leverages the structural and functional similarities within protein families. Two distinct but complementary experimental approaches have emerged within this framework: forward chemogenomics and reverse chemogenomics. These paradigms differ fundamentally in their starting points and methodologies while sharing the ultimate goal of identifying novel therapeutic agents and their mechanisms of action.
Forward Chemogenomics: From Phenotype to Target
Conceptual Foundation and Workflow
Forward chemogenomics, also termed "classical chemogenomics," begins with the observation of a desired phenotype in a cell or organism and works backward to identify the molecular targets responsible [1] [2]. This phenotype-first approach involves screening compound libraries against intact biological systems to identify molecules that induce a specific phenotypic change, followed by deconvolution efforts to determine the protein target and mechanism of action underlying the observed phenotype [1]. The fundamental strength of this strategy lies in its unbiased nature—it does not require presupposed knowledge of specific molecular targets, making it particularly valuable for investigating complex biological processes and polygenic diseases where the key molecular players may be unknown.
In forward chemogenomics, the conditional effects of chemical compounds on entire biological systems are measured, allowing researchers to identify active chemicals based on their phenotypic influence rather than their inhibition of a specific protein target [2]. This approach mirrors traditional phenotypic screening but enhances it with modern genomic technologies and computational target identification methods. The main challenge lies in designing phenotypic assays that enable a relatively straightforward path from screening to target identification, which often requires sophisticated genetic, biochemical, or computational deconvolution strategies [1].
Table: Key Characteristics of Forward Chemogenomics
| Aspect | Description |
|---|---|
| Starting Point | Phenotypic observation in biological system [1] |
| Screening Focus | Conditional effects of compounds on entire biological systems [2] |
| Target Knowledge | Molecular basis of phenotype initially unknown [1] |
| Primary Challenge | Target identification and deconvolution [1] |
| Strength | Unbiased discovery without target presupposition [1] |
Experimental Methodologies and Protocols
The experimental workflow for forward chemogenomics typically begins with establishing a robust phenotypic assay that accurately captures a disease-relevant biological process. Advanced technologies have significantly enhanced the power and scalability of this approach. High-content imaging and single-cell technologies now enable the capture of subtle, disease-relevant phenotypes at scale [3]. Modern implementations often employ multiplexed assays, single-cell sequencing, and automated imaging to generate multi-dimensional phenotypic profiles [3].
Recent methodological innovations include pooled perturbations with computational deconvolution, which dramatically reduce sample size, labor, and cost while maintaining information-rich outputs [3]. For example, compressed phenotypic screening using pooled perturbations allows researchers to screen multiple conditions simultaneously and computationally deconvolve the results, enabling the testing of thousands of genetic or chemical perturbations in a single experiment [3]. These approaches are further powered by AI and machine learning models that interpret massive, noisy datasets to detect meaningful patterns that might escape human observation [3].
The target identification phase typically employs one of several established methodologies:
- Affinity purification: Using the active compound as bait to pull down interacting proteins from cell lysates.
- Genetic approaches: Screening for resistance mutations or synthetic lethal interactions that point to potential targets.
- Transcriptomic profiling: Comparing gene expression patterns induced by the compound to reference databases.
- Proteomic profiling: Using techniques like thermal shift assays to identify proteins whose stability changes upon compound binding.
Each of these methods has strengths and limitations, and often multiple approaches are combined to confidently identify the molecular target responsible for the observed phenotype.
Reverse Chemogenomics: From Target to Phenotype
Conceptual Foundation and Workflow
Reverse chemogenomics represents the complementary approach to forward chemogenomics, beginning with a specific molecular target of interest and proceeding to identify compounds that modulate its activity, then determining the phenotypic consequences of this modulation [1]. This target-first strategy expresses gene sequences of interest as target proteins and screens chemical libraries in a high-throughput, target-based manner [2]. The reverse approach essentially formalizes and enhances the target-based drug discovery strategies that have dominated pharmaceutical research in recent decades, with the key distinction being its emphasis on parallel screening across entire gene and protein families based on structure-activity relationship homology concepts [2].
In this paradigm, small compounds that perturb the function of a specific target protein are first identified through in vitro biochemical assays, and the phenotypic effects of these active compounds are subsequently analyzed in cellular or whole-organism models [1]. This strategy is particularly powerful when there is strong genetic or biological evidence implicating a specific molecular target in a disease process, allowing for a more focused discovery approach. Reverse chemogenomics benefits from well-established screening technologies and typically offers a more straightforward path from hit identification to lead optimization, as the molecular target is known from the outset.
Table: Key Characteristics of Reverse Chemogenomics
| Aspect | Description |
|---|---|
| Starting Point | Known molecular target with suspected therapeutic relevance [1] |
| Screening Focus | High-throughput target-based screening of chemical libraries [2] |
| Target Knowledge | Target identity and function known from outset [1] |
| Primary Challenge | Establishing physiological relevance and phenotypic impact [1] |
| Strength | Streamlined path from hit to lead with known mechanism [1] |
Experimental Methodologies and Protocols
The reverse chemogenomics workflow typically begins with the selection and production of the target protein, often focusing on specific protein families with known therapeutic relevance (e.g., kinases, GPCRs, ion channels). Target proteins are expressed and purified, followed by the development of robust biochemical assays that can be scaled for high-throughput screening. These assays are designed to measure direct compound-target interactions, typically using techniques such as fluorescence-based activity assays, binding assays, or structural biology approaches.
Modern implementations of reverse chemogenomics leverage advanced computational and structural biology methods to enhance efficiency. For example, AI-driven platforms can predict drug-target binding affinities using multitask deep learning frameworks that learn the structural properties of drug molecules, the conformational dynamics of proteins, and the bioactivity between drugs and targets [4]. These computational approaches can significantly accelerate the initial screening phase by prioritizing compounds with a higher likelihood of interaction.
After identifying target-active compounds, researchers progress to phenotypic validation in cellular and organismal models. This critical step determines whether modulation of the target produces the desired therapeutic effect and helps identify potential mechanism-based toxicities. Contemporary approaches often incorporate multi-omics profiling to comprehensively characterize the downstream effects of target modulation, including transcriptomic, proteomic, and metabolomic changes [3].
Key methodological considerations in reverse chemogenomics include:
- Assay design: Developing physiologically relevant assays that capture the target's native conformation and function.
- Library design: Creating focused libraries enriched with compounds likely to interact with the target family.
- Selectivity profiling: Assessing compound activity across related targets to achieve desired selectivity profiles.
- Structural biology: Using X-ray crystallography or cryo-EM to guide compound optimization through structure-based drug design.
Comparative Analysis: Strategic Applications and Limitations
Side-by-Side Paradigm Comparison
Forward and reverse chemogenomics represent complementary strategies with distinct advantages and limitations that make them suitable for different research contexts. The following table provides a comprehensive comparison of these two approaches across multiple dimensions:
Table: Comprehensive Comparison of Forward and Reverse Chemogenomics
| Dimension | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotypic observation in complex biological system [1] | Defined molecular target with suspected disease relevance [1] |
| Screening Context | Intact cells or organisms [2] | Isolated molecular targets [2] |
| Target Knowledge | Initially unknown; identified through deconvolution [1] | Known from outset [1] |
| Primary Strength | Unbiased discovery; identification of novel targets and mechanisms [1] | Streamlined optimization; clear mechanism of action [1] |
| Primary Challenge | Target identification and validation [1] | Establishing physiological relevance and phenotypic impact [1] |
| Typical Applications | Complex diseases, polygenic disorders, pathway discovery [1] | Well-validated target classes, structure-based drug design [1] |
| Technical Requirements | Phenotypic assays, target deconvolution platforms [3] | Protein production, high-throughput screening automation [2] |
| Success Rate | Higher likelihood of phenotypic efficacy but longer timeline to target | Faster to lead optimization but potential translational failures |
| Data Output | Multi-dimensional phenotypic profiles [3] | Structure-activity relationships, binding affinities [4] |
Strategic Implementation in Drug Discovery
The choice between forward and reverse chemogenomics depends heavily on the specific research context, available tools, and stage of discovery. Forward chemogenomics excels in situations where the molecular pathophysiology of a disease is poorly understood but robust phenotypic assays exist. It has proven particularly valuable for identifying novel targets in complex diseases such as cancer, neurodegenerative disorders, and infectious diseases, where multiple redundant pathways may be involved [3] [1]. The resurgence of phenotypic screening, powered by modern omics data and AI, signals a shift back to this biology-first approach, which can uncover therapeutic opportunities that target-centric methods might miss [3].
Reverse chemogenomics demonstrates particular strength when a specific target family has been genetically or clinically validated in a disease process. This approach enables efficient exploration of chemical space against well-characterized target classes, leveraging accumulated knowledge about structure-activity relationships within these families [1]. The parallel screening of compound libraries across multiple members of a target family facilitates the rapid identification of selective modulators while understanding potential off-target effects from the outset. Modern AI-driven platforms have enhanced this approach through capabilities such as drug-target affinity prediction and target-aware drug generation using multitask deep learning frameworks [4].
In contemporary drug discovery, the most successful organizations strategically integrate both approaches, recognizing their complementary nature. Many discovery pipelines begin with forward chemogenomics to identify novel targets and mechanisms, then transition to reverse chemogenomics approaches for lead optimization and portfolio expansion around validated targets. This integrated strategy leverages the strengths of both paradigms while mitigating their respective limitations.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of chemogenomic approaches requires specialized reagents and tools designed to address the unique challenges of systematic chemical-genetic interaction mapping. The following table details essential research solutions for chemogenomics investigations:
Table: Essential Research Reagent Solutions for Chemogenomics
| Reagent/Tool | Function | Application Context |
|---|---|---|
| Targeted Chemical Libraries | Focused compound collections enriched for specific protein families (kinases, GPCRs, etc.) [1] | Both forward and reverse chemogenomics; enables efficient screening of target families |
| Cell Painting Assays | High-content imaging using fluorescent dyes to visualize multiple organelles [3] | Forward chemogenomics; generates rich morphological profiles for phenotypic screening |
| Perturb-seq Technologies | Pooled CRISPR screens with single-cell RNA sequencing readout [3] | Forward chemogenomics; enables large-scale genetic perturbation studies |
| High-Content Screening Systems | Automated microscopy and image analysis platforms [3] | Forward chemogenomics; quantitative phenotypic analysis at scale |
| Drug-Target Affinity Prediction Models | AI models predicting binding strengths between compounds and targets [4] | Reverse chemogenomics; prioritizes compounds for experimental testing |
| Target-Aware Drug Generation Systems | Generative AI models designing novel compounds for specific targets [4] | Reverse chemogenomics; creates novel chemical matter for target families |
| Multi-omics Profiling Platforms | Integrated genomic, transcriptomic, proteomic, and metabolomic analyses [3] | Both paradigms; provides systems-level view of compound effects |
| Knowledge Graph Tools | Computational networks integrating biological relationships for target deconvolution [5] | Forward chemogenomics; identifies molecular targets from phenotypic hits |
Forward and reverse chemogenomics represent complementary paradigms that collectively enable comprehensive exploration of the chemical-biological interface. While forward chemogenomics begins with phenotypic observations and progresses to target identification, reverse chemogenomics starts with defined molecular targets and assesses phenotypic consequences [1] [2]. Both approaches have been strengthened by technological advancements in high-throughput screening, omics technologies, and computational methods, particularly artificial intelligence and machine learning.
The integration of these approaches creates a powerful drug discovery engine capable of both de novo target discovery and efficient lead optimization. Modern AI-driven platforms exemplify this integration, leveraging multimodal data fusion to build comprehensive biological representations that inform both target identification and compound design [5]. As these technologies continue to evolve, the distinction between forward and reverse chemogenomics may increasingly blur, giving way to integrated systems that simultaneously optimize chemical and biological understanding in a continuous feedback loop.
The future of chemogenomics lies in further strengthening this integration, with advances in single-cell technologies, structural biology, and artificial intelligence promising to enhance both the scale and precision of chemical-genetic interaction mapping. By strategically employing both forward and reverse paradigms, researchers can accelerate the discovery of novel therapeutic agents while deepening our fundamental understanding of biological systems.
In the post-genomic era, chemogenomics has emerged as a systematic approach for screening targeted chemical libraries against families of drug targets with the ultimate goal of identifying novel drugs and drug targets simultaneously [1]. This field represents a fundamental integration of target and drug discovery by using active compounds as probes to characterize proteome functions [1]. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention, and chemogenomics strategically addresses the intersection of all possible drugs on all these potential targets [1]. The core strategic workflows in this field are categorized into two distinct but complementary approaches: forward chemogenomics and reverse chemogenomics [6] [1]. Both approaches require suitable compound collections and appropriate model systems for screening, with the biologically active compounds discovered through these methods serving as "modulators" that bind to and modulate specific molecular targets, making them valuable as potential targeted therapeutics [1]. This technical guide examines these two foundational pathways, their methodological frameworks, applications, and integration within modern drug discovery pipelines.
Forward Chemogenomics: From Phenotype to Target
Conceptual Framework and Workflow
Forward chemogenomics, also termed classical chemogenomics, begins with the investigation of a particular phenotype of interest, followed by the identification of small molecules that interact with this function [1]. The key differentiator of this approach is that the molecular basis of the desired phenotype is initially unknown [1]. For example, researchers might begin with a loss-of-function phenotype such as the arrest of tumor growth, and then identify compounds that induce this target phenotype [1]. Once these modulators are identified, they serve as tools to investigate the protein responsible for the phenotype [1]. The primary challenge in forward chemogenomics lies in designing phenotypic assays that can efficiently transition from screening to target identification [1].
The following workflow diagram illustrates the strategic pathway of forward chemogenomics:
Experimental Methodologies and Protocols
The implementation of forward chemogenomics requires specialized experimental protocols designed to connect phenotypic observations to molecular targets:
Phenotypic Assay Development: Design cell-based or whole-organism assays that accurately recapitulate the disease-relevant phenotype. These assays typically utilize high-content screening systems that monitor multiple parameters such as cell morphology, proliferation, death, or specific reporter gene expression [7]. For example, an anti-cancer phenotypic screen might use 3D tumor spheroids or patient-derived organoids to identify compounds that inhibit growth while maintaining viability of non-cancerous cells [7].
High-Throughput Phenotypic Screening: Screen diverse compound libraries using automated systems. The EUbOPEN consortium, for instance, has developed chemogenomic libraries covering approximately one-third of the druggable proteome, which are particularly valuable for such phenotypic screens [8]. These libraries include compounds with well-characterized but overlapping target profiles, enabling target deconvolution based on selectivity patterns observed in phenotypic assays [8].
Target Deconvolution Techniques: Once bioactive compounds are identified, several methods can be employed to identify their molecular targets:
- Affinity Purification: Immobilize hit compounds on solid supports and use them as bait to pull down interacting proteins from cell lysates, followed by mass spectrometric identification.
- Chemoproteomics: Use chemical probes derived from hit compounds to capture protein targets directly in complex proteomes.
- Resistance Mutagenesis: Generate resistant cell lines through chemical mutagenesis and identify mutated genes through whole-exome sequencing.
- Transcriptomic Profiling: Compare gene expression signatures of hit compounds to reference compounds with known mechanisms.
Validation Studies: Confirm target engagement and functional relevance using orthogonal approaches such as CRISPR-Cas9 gene editing, RNA interference, or biophysical methods like surface plasmon resonance to measure direct binding affinities [8].
Reverse Chemogenomics: From Target to Phenotype
Conceptual Framework and Workflow
Reverse chemogenomics represents the complementary approach to forward chemogenomics, beginning with a defined molecular target rather than an observed phenotype [1]. In this strategy, small compounds that perturb the function of a specific protein (such as an enzyme or receptor) are first identified through in vitro biochemical assays [1]. After modulators are identified, the phenotype induced by these molecules is analyzed in cellular systems or whole organisms [1]. This method serves to identify or confirm the biological function of the protein and its potential therapeutic relevance [1]. While this approach shares similarities with traditional target-based drug discovery, it is enhanced by parallel screening capabilities and the ability to perform lead optimization across multiple targets belonging to the same gene family [1].
The strategic pathway for reverse chemogenomics is systematically outlined in the following workflow:
Experimental Methodologies and Protocols
Reverse chemogenomics employs target-centric experimental protocols that progress from molecular interactions to systems-level phenotypes:
Target Selection and Validation: Select biologically relevant targets based on genomic, genetic, or clinical evidence. The EUbOPEN consortium focuses particularly on understudied target families such as E3 ubiquitin ligases and solute carriers (SLCs) to expand the druggable proteome [8]. Target validation may include analysis of disease-associated genetic variants, protein expression patterns in pathological states, or functional evidence from model organisms.
Biochemical Assay Development: Develop robust high-throughput screening assays that measure compound effects on target activity. For enzymes, this typically involves fluorescence-based, luminescence-based, or absorbance-based readouts of catalytic activity. For receptors, binding assays using labeled ligands or functional assays measuring downstream signaling events are employed. The EUbOPEN consortium has established strict criteria for chemical probes, including potency <100 nM in in vitro assays and selectivity of at least 30-fold over related proteins [8].
Target-Based Screening: Screen compound libraries against the purified target protein or simplified cellular systems. The EUbOPEN project utilizes chemogenomic compound sets where compounds may bind multiple targets but have well-characterized selectivity profiles [8]. These compounds are valuable tools for reverse chemogenomics as their overlapping target profiles facilitate the interpretation of phenotypic outcomes.
Cellular Phenotype Characterization: Evaluate the functional consequences of target modulation in relevant cellular models. This includes assessment of pathway modulation (e.g., phosphorylation status, second messenger levels), gene expression changes, and phenotypic effects such as proliferation, differentiation, or death. For more physiologically relevant models, 3D culture systems or patient-derived cells are increasingly utilized [7].
In Vivo Validation: Confirm therapeutic potential in animal models of disease. This step establishes whether pharmacological modulation of the target produces the desired therapeutic effect while maintaining an acceptable safety profile. The EUbOPEN consortium profiles compounds in patient-derived disease assays, with particular focus on inflammatory bowel disease, cancer, and neurodegeneration [8].
Comparative Analysis: Strategic Implementation
Side-by-Side Workflow Comparison
The strategic differences between forward and reverse chemogenomics can be visualized through their integrated workflow, highlighting their complementary nature:
Quantitative Comparison of Strategic Approaches
The following table provides a systematic comparison of the technical and strategic specifications for both chemogenomics approaches:
| Parameter | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotypic observation in cells or organisms [1] | Known or hypothesized molecular target [1] |
| Primary Screening Method | Phenotypic assays (high-content screening) [1] | Target-based assays (biochemical, binding) [1] |
| Hit Identification Criteria | Compounds inducing desired phenotype [1] | Compounds modulating target activity [1] |
| Key Challenge | Target deconvolution [1] | Phenotypic relevance [1] |
| Typical Timeline | Longer (due to target identification phase) | Shorter (focused target approach) |
| Risk Factors | Difficulty identifying molecular target; off-target effects | Poor translation from in vitro to in vivo efficacy |
| Major Advantage | Unbiased discovery; identification of novel biology | Streamlined optimization; clearer mechanism |
| Data Integration Needs | Multi-omics data for target validation | Structural biology and cheminformatics for optimization |
| Automation Potential | Moderate (complex phenotypes harder to automate) | High (standardized biochemical assays) |
Applications and Case Studies
Both forward and reverse chemogenomics have demonstrated significant utility across various applications in drug discovery and biological research:
Mode of Action Determination: Chemogenomics has been successfully applied to determine the mode of action for traditional medicines, including Traditional Chinese Medicine and Ayurveda [1]. For example, computational analysis of compounds in "toning and replenishing medicine" from TCM identified sodium-glucose transport proteins and PTP1B as targets relevant to the hypoglycemic phenotype, providing mechanistic insights into traditional remedies [1].
Novel Target Identification: Reverse chemogenomics profiling enabled the identification of new antibacterial targets by capitalizing on existing ligand libraries for the murD enzyme in the peptidoglycan synthesis pathway [1]. Researchers applied the chemogenomics similarity principle to map murD ligands to other members of the mur ligase family (murC, murE, murF, murA, and murG), identifying new targets for known ligands that could serve as broad-spectrum Gram-negative inhibitors [1].
Pathway Gene Discovery: Forward chemogenomics approaches using yeast cofitness data identified YLR143W as the previously unknown diphthamide synthetase enzyme, solving a 30-year mystery in the final step of diphthamide biosynthesis [1]. By identifying strains with high cofitness to known diphthamide biosynthesis genes, researchers successfully pinpointed the missing enzyme in the pathway [1].
Chemical Probe Development: The EUbOPEN consortium has developed rigorous criteria for chemical probes, including potency <100 nM in in vitro assays, selectivity ≥30-fold over related proteins, evidence of target engagement in cells at <1 μM, and a reasonable cellular toxicity window [8]. These probes serve as critical tools for both forward and reverse chemogenomics approaches.
Essential Research Reagents and Tools
The Scientist's Toolkit: Core Research Reagents
Successful implementation of chemogenomics approaches requires specialized reagents and tools. The following table outlines essential solutions and their applications:
| Research Reagent | Function & Application | Examples/Specifications |
|---|---|---|
| Chemogenomic Compound Libraries | Collections of compounds with known activity against target families; used for both forward and reverse screening [8] | EUbOPEN library (covers 1/3 of druggable proteome); kinase inhibitor sets; GPCR ligand libraries [8] |
| Chemical Probes | Highly characterized, potent, and selective small molecules for specific target modulation [8] | Potency <100 nM; ≥30-fold selectivity; cellular activity <1 μM; with matched negative controls [8] |
| Phenotypic Screening Assays | Cell-based or organoid models for detecting phenotypic changes in forward chemogenomics [7] | High-content imaging assays; 3D culture systems; patient-derived primary cells [7] |
| Target-Based Assay Systems | Biochemical platforms for measuring compound effects on specific targets in reverse chemogenomics | Fluorescence polarization; TR-FRET; enzymatic activity assays; binding assays |
| Chemoproteomic Platforms | Tools for target deconvolution in forward chemogenomics | Affinity chromatography matrices; activity-based probes; photoaffinity labeling reagents |
| Data Curation Tools | Software for ensuring chemical and biological data quality [9] | Molecular standardization tools; duplicate detection algorithms; bioactivity outlier filters [9] |
Integration and Future Directions
The strategic integration of forward and reverse chemogenomics creates a powerful iterative cycle for drug discovery. As the EUbOPEN consortium demonstrates, the systematic generation of chemogenomic compound sets and high-quality chemical probes enables both target-centered and phenotype-driven approaches to converge on validated therapeutic strategies [8]. The field is further enhanced by computational approaches, including machine learning methods that predict drug-target interactions and optimize molecular properties [6] [10]. Deep learning architectures, such as convolutional neural networks and recurrent neural networks, have shown particular utility in predicting molecular properties, protein structures, and ligand-target interactions, thereby accelerating lead compound identification and optimization [10].
Future developments in chemogenomics will likely focus on expanding the druggable proteome through new modalities such as molecular glues, PROTACs, and other proximity-inducing small molecules [8]. The EUbOPEN consortium has already begun focusing on challenging target classes including E3 ubiquitin ligases and solute carriers, pushing the field to evolve criteria for new modalities including covalent binders and PROTACs [8]. As these tools and approaches mature, the strategic workflow integrating forward and reverse chemogenomics will continue to provide a systematic framework for translating genomic information into therapeutic breakthroughs.
This technical guide was developed referencing current literature and consortium guidelines, including the EUbOPEN project and Target 2035 initiative, which aim to generate chemical modulators for nearly all human proteins by 2035 [8].
Historical Context and Evolution in Modern Drug Discovery
Modern drug discovery has undergone a profound transformation, evolving from a largely serendipitous process to a systematic, data-driven science. This evolution has been catalyzed by the completion of the human genome project, which provided an abundance of potential therapeutic targets, and by advances in chemical biology that enabled the systematic screening of small molecules against these targets [1]. Chemogenomics, or chemical genomics, represents this modern paradigm, defined as the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identifying novel drugs and drug targets [1]. This approach strategically integrates target and drug discovery by using active compounds as probes to characterize proteome functions, allowing researchers to study the intersection of all possible drugs on all potential targets [1].
The expensive and time-consuming nature of traditional drug discovery is no longer feasible, with estimates indicating an average cost of $2.6 billion and timelines exceeding 12 years for a complete traditional workflow [7]. In response to these challenges, computational methods, particularly computer-aided drug discovery (CADD), have revolutionized the field by providing cost-efficient ways to reduce failures for high-throughput screening, produce new ideas for rational drug design, and rationally anticipate targeted protein and candidate hits [11]. These advances have crystallized into two fundamental methodological frameworks: forward chemogenomics and reverse chemogenomics, which provide complementary pathways for interrogating biological systems and identifying therapeutic interventions.
Historical Foundations: From Classical Approaches to Chemogenomics
The Pre-Chemogenomics Era
Historically, drug discovery relied heavily on natural products, with knowledge of toxic or medicinal properties often long predating understanding of precise targets or mechanisms [12]. Natural selection provided a slow but steady stream of bioactive small molecules, but each needed to confer reproductive advantage for nature to 'invest' in its synthesis [12]. The revolution in molecular biology shifted screening toward purified proteins, but with advances in assay technology, research programs increasingly returned to cell- or organism-based phenotypic assays that preserve cellular context [12].
The conceptual framework for modern chemogenomics emerged by analogy to genetics. Forward genetics identifies phenotypes of interest first, followed by identification of responsible genes, while reverse genetics targets specific genes of interest first, then searches for resulting phenotypes [12]. Similarly, the two fundamental approaches to understanding small molecule action on biological systems became known as forward and reverse chemical genetics [12].
The Rise of Computational Approaches
In 1981, an influential article titled "Next Industrial Revolution: Designing Drugs by Computer at Merck" marked a turning point in recognizing the importance of in silico studies in drug discovery [11]. Since then, high-throughput screening (HTS) has been increasingly used in pharmaceutical and academic institutions to rapidly discover hit and lead compounds [11]. The development of virtual high-throughput screening (vHTS) addressed limitations of traditional HTS by using virtual compound libraries, allowing experimentalists to focus on ligands more likely to have activity of interest [11]. This computational revolution provided the essential infrastructure for modern chemogenomics approaches.
Forward vs. Reverse Chemogenomics: Conceptual Frameworks and Workflows
Forward Chemogenomics
Forward chemogenomics (also known as classical chemogenomics) begins with the investigation of a particular phenotype, where the molecular basis is unknown [1]. Researchers identify small compounds that interact with this function, then use these modulators as tools to identify the protein responsible for the phenotype [1]. For example, a loss-of-function phenotype could be an arrest of tumor growth, and once compounds that lead to this target phenotype are identified, the next step involves identifying the responsible genes and protein targets [1].
The main challenge of forward chemogenomics strategy lies in designing phenotypic assays that lead immediately from screening to target identification [1]. This approach benefits from preserving cellular context and can discover new therapeutic targets without preconceived notions of relevant targets and signaling pathways [12].
Forward Chemogenomics Workflow:
Reverse Chemogenomics
Reverse chemogenomics takes the opposite approach, beginning with small compounds that perturb the function of an enzyme in the context of an in vitro enzymatic test [1]. Once modulators are identified, the phenotype induced by the molecule is analyzed in cellular tests or whole organisms [1]. This method confirms the role of the enzyme in the biological response and was historically virtually identical to target-based approaches applied in drug discovery and molecular pharmacology [1].
This strategy has been enhanced by parallel screening and the ability to perform lead optimization on many targets belonging to one target family [1]. Reverse chemogenomics benefits from clearer initial target validation but may miss complex cellular contexts that affect drug action [12].
Reverse Chemogenomics Workflow:
Comparative Analysis: Forward vs. Reverse Chemogenomics
Table 1: Comparison of Forward and Reverse Chemogenomics Approaches
| Parameter | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotype of interest | Known protein target |
| Screening Context | Cells or whole organisms | Purified proteins or simplified systems |
| Target Identification | Required after compound identification | Known from the beginning |
| Advantages | Discovers novel targets and pathways; preserves biological context | Clear target validation; streamlined for known target families |
| Challenges | Difficult target deconvolution; complex data interpretation | May miss relevant cellular context; limited to known targets |
| Typical Applications | Phenotypic drug discovery; mechanism of action studies | Targeted drug development; lead optimization |
| Historical Examples | Cyclosporine A/FK506 discovery of FKBP12, calcineurin, mTOR [12] | Kinase inhibitor development; protease-targeted drugs |
Computational Methodologies in Chemogenomics
Reverse Screening for Target Identification
A critical challenge in forward chemogenomics is identifying protein targets after phenotypic screening. Reverse screening methods have been developed to address this need, with three major computational approaches emerging [13]:
Shape Screening: Identifies potential targets by comparing the overall shape of a query molecule to ligands in annotated databases. The basic principle is that structurally similar molecules may have similar bioactivity by targeting the same proteins [13].
Pharmacophore Screening: Compares key pharmacophore features (specific arrangements of chemical features essential for biological activity) rather than overall shape, using databases annotated with target information [13].
Reverse Docking: Successively docks a query molecule into the active pocket of each protein in a 3D structure database based on spatial and energy principles to identify protein targets with strong binding affinity [13].
Table 2: Computational Tools for Reverse Screening in Chemogenomics
| Method | Representative Tools | Key Databases | Applications |
|---|---|---|---|
| Shape Screening | ChemMapper, SEA, TargetHunter | ChEMBL, PubChem, BindingDB | Initial target hypothesis generation; polypharmacology prediction |
| Pharmacophore Screening | PharmMapper, Pharao | IUPHAR, PDSP Ki Database | Mechanism of action studies; off-target effect prediction |
| Reverse Docking | INVDOCK, idTarget | Protein Data Bank (PDB) | Structure-based target identification; binding site analysis |
Advanced Computational Frameworks
Recent advances have introduced multitask learning frameworks that simultaneously predict drug-target interactions and generate novel target-aware drug candidates. The DeepDTAGen framework represents a cutting-edge example, utilizing shared feature spaces for both predicting drug-target binding affinities and generating new drug variants [4]. This approach addresses optimization challenges in multitask learning through novel algorithms like FetterGrad, which mitigates gradient conflicts between distinct tasks [4].
The emergence of the "informacophore" concept further extends traditional pharmacophore models by incorporating computed molecular descriptors, fingerprints, and machine-learned representations of chemical structure [7]. This data-driven approach identifies minimal chemical structures combined with computational descriptors essential for biological activity, enabling more systematic and bias-resistant strategies for scaffold modification and optimization [7].
Experimental Protocols in Modern Chemogenomics
Target Identification Protocols
Protocol 1: Limited Proteolysis Coupled to Mass Spectrometry (LiP-MS) for Target Deconvolution
This direct biochemical method identifies protein targets through structural proteomics [14]:
- Cell Lysis: Prepare native cell lysates from relevant cell lines under non-denaturing conditions.
- Compound Treatment: Incubate lysates with small molecule of interest or DMSO control.
- Limited Proteolysis: Subject samples to limited proteolysis with nonspecific protease (e.g., proteinase K).
- Protease Inactivation: Denature proteins and inactivate protease.
- Tryptic Digest: Digest samples with trypsin for mass spectrometry analysis.
- LC-MS/MS Analysis: Analyze peptides by liquid chromatography coupled to tandem mass spectrometry.
- Data Analysis: Identify protein targets by detecting proteolytic patterns that change upon compound binding.
Protocol 2: Cellular Thermal Shift Assay (CETSA) for Target Engagement
CETSA validates target engagement in cellular contexts by detecting thermal stabilization of proteins upon ligand binding [14]:
- Cell Treatment: Treat cells with compound of interest or vehicle control.
- Heat Challenge: Subject aliquots of cell suspension to different temperatures.
- Cell Lysis: Lyse cells and separate soluble protein fraction.
- Protein Quantification: Detect remaining soluble target protein by immunoblotting or quantitative MS.
- Data Analysis: Calculate thermal shift (ΔTm) to confirm compound binding.
Phenotypic Screening Protocols
Protocol 3: 3D Spheroid Invasion Assay for High-Throughput Screening
This phenotypic assay models cancer cell invasion and response to compounds in a more physiologically relevant 3D context [14]:
- Spheroid Formation: Seed cells in ultra-low attachment plates to form uniform spheroids.
- Matrix Embedding: Embed spheroids in extracellular matrix (e.g., Matrigel) to simulate tissue environment.
- Compound Treatment: Treat spheroids with chemogenomic library compounds.
- Live-Cell Imaging: Monitor spheroid invasion over 3-5 days using automated microscopy.
- Image Analysis: Quantify invasion area using segmentation algorithms (e.g., Ilastik, ImageJ).
- Hit Identification: Identify compounds that significantly inhibit invasion compared to controls.
Protocol 4: High-Content Live-Cell Imaging for Cell Health Profiling
This multiparametric assay simultaneously evaluates multiple cell health parameters in response to compound treatment [14]:
- Cell Seeding: Seed cells in multi-well plates optimized for imaging.
- Compound Treatment: Treat with chemogenomic library compounds across concentration range.
- Live-Cell Staining: Incubate with fluorescent dyes for viability, apoptosis, cell cycle, and organelle health.
- Automated Imaging: Acquire images using high-content microscope with environmental control.
- Multiparametric Analysis: Extract features for each cell using image analysis software (e.g., CellPathfinder).
- Phenotypic Profiling: Cluster compounds based on multiparametric responses to identify mechanisms.
Essential Research Reagents and Solutions
Table 3: Research Reagent Solutions for Chemogenomics Experiments
| Reagent/Solution | Function | Example Applications |
|---|---|---|
| Kinase Chemogenomic Set (KCGS) | Targeted library covering kinase families with well-annotated inhibitors | Kinase target validation; polypharmacology profiling |
| NanoLuc Binary Technology (NanoBRET) | Bioluminescence resonance energy transfer system for monitoring protein-protein interactions | Live-cell target engagement studies; kinase selectivity profiling |
| HiBiT Tagging System | Small peptide tag (11 amino acids) for highly sensitive protein detection | Cellular Thermal Shift Assay (CETSA); protein stability monitoring |
| Photoaffinity Probes | Chemically modified compounds with photoreactive groups for covalent target capture | Target identification for phenotypic screening hits |
| Functional Assay Kits | Pre-optimized reagent sets for specific pathway readouts (apoptosis, autophagy, etc.) | Mechanism of action studies; pathway validation |
Current Research and Emerging Applications
Multitask Deep Learning in Drug-Target Interaction Prediction
The DeepDTAGen framework exemplifies modern computational approaches, performing both drug-target affinity prediction and target-aware drug generation simultaneously using common features [4]. This model addresses the traditionally separate tasks of predictive modeling (identifying interactions) and generative modeling (designing new drugs) through a unified architecture. Comprehensive experiments on benchmark datasets (KIBA, Davis, BindingDB) demonstrate robust performance, with the model achieving MSE of 0.146, CI of 0.897, and r²m of 0.765 on KIBA test sets [4].
Integration with Traditional Medicine
Chemogenomics has been applied to identify mode of action for traditional medicinal systems, including Traditional Chinese Medicine (TCM) and Ayurveda [1]. Compounds from traditional medicines are often more soluble than synthetic compounds, have "privileged structures" frequently found to bind in different living organisms, and have better-characterized safety profiles [1]. In silico analysis using target prediction programs has helped identify target-phenotype links for traditional medicines, such as connecting sodium-glucose transport proteins and PTP1B to the hypoglycemic phenotype of "toning and replenishing medicine" in TCM [1].
Combination Chemical Genetics
Combination chemical genetics (CCG) extends basic chemogenomics principles by systematically applying multiple chemical or mixed chemical and genetic perturbations [15]. This approach helps identify functional relationships between pathways and component modules that aren't apparent from single perturbations [15]. CCG is particularly valuable for identifying synthetic lethal interactions in cancer therapy and understanding network-level responses to complex perturbations [15].
The evolution of modern drug discovery from serendipitous finding to systematic chemogenomics represents a paradigm shift in how we approach therapeutic development. Forward and reverse chemogenomics provide complementary frameworks that integrate target and drug discovery, accelerated by computational methods and high-throughput experimental technologies. The convergence of large-scale chemical libraries, advanced screening technologies, and sophisticated computational approaches including machine learning and multitask deep learning continues to reshape the landscape of drug discovery.
Future directions will likely involve greater integration of artificial intelligence throughout the discovery pipeline, increased use of physiologically relevant model systems (such as organoids and organs-on-chips), and more sophisticated multi-omics integration for comprehensive compound profiling. The distinction between forward and reverse approaches will continue to blur as integrated platforms emerge that simultaneously address target identification, compound optimization, and mechanism elucidation. As these technologies mature, chemogenomics will solidify its position as the foundational framework for 21st-century therapeutic discovery, enabling more efficient, targeted, and successful development of novel medicines for human disease.
Phenotypic Screening, Target-Based Assays, and Chemogenomic Space
The journey of drug discovery has evolved from a largely serendipitous endeavor to a sophisticated, multi-faceted scientific discipline. At the heart of this evolution lies the tension between two fundamental approaches: phenotypic screening, which identifies compounds based on their observable effects in complex biological systems, and target-based screening, which seeks compounds that modulate specific, predefined molecular targets [16] [17]. Historically, phenotypic screening was the foundation for most drug discovery, with the molecular mechanism of action (MMOA) often determined years after a drug's therapeutic effect was observed—a process known as "classical pharmacology" or "forward pharmacology" [17]. The late 20th century saw a major shift toward target-based approaches, fueled by advances in genomics and molecular biology that promised more rational and efficient discovery [18] [17].
However, a pivotal analysis revealed a shortcoming of the target-based paradigm: phenotypic screening has been the more successful strategy for discovering first-in-class medicines with novel mechanisms of action [18] [17]. This discovery has spurred a renaissance for phenotypic methods, albeit now integrated with modern tools and technologies. Bridging these two worlds is the emerging discipline of chemogenomics, which systematically explores the interaction between chemical libraries and target families on a genome-wide scale [1]. Chemogenomics provides a conceptual and experimental framework—the "chemogenomic space"—to navigate the intersection of all possible drugs and all potential targets [1]. This guide will delve into the core concepts of phenotypic screening, target-based assays, and chemogenomic space, framing them within the critical distinction between forward and reverse chemogenomics research.
Conceptual Framework: Forward vs. Reverse Chemogenomics
Chemogenomics aims to systematically identify novel drugs and drug targets by screening targeted chemical libraries against specific families of drug targets, such as G-protein-coupled receptors (GPCRs) or kinases [1]. It integrates target and drug discovery by using small molecules as probes to characterize biological function. This field is fundamentally divided into two complementary experimental approaches.
Forward Chemogenomics
Forward chemogenomics, also termed classical chemogenomics, begins with an observed phenotype. Researchers screen for small molecules that induce a desired phenotypic change in a cell or organism, such as the arrest of tumor growth, without any prior assumption about the molecular target [1] [19]. The primary challenge lies in the subsequent target deconvolution—identifying the protein target and molecular pathway responsible for the observed phenotype [1]. This approach is unbiased and has been instrumental in discovering first-in-class therapies [18].
Reverse Chemogenomics
Reverse chemogenomics starts with a defined molecular target. It identifies small molecules that perturb the function of a specific protein (e.g., in an in vitro enzymatic assay) and then analyzes the phenotypic consequences of this interaction in cells or whole organisms [1]. This strategy, which closely mirrors traditional target-based drug discovery, is powerful for validating a hypothesis that a specific target is disease-modifying [1] [17]. It has been enhanced by parallel screening and the ability to optimize compounds across entire target families.
Table 1: Comparison of Forward and Reverse Chemogenomics
| Feature | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Desired phenotype in a complex system [1] | Defined protein target [1] |
| Primary Screening | Phenotypic assay (e.g., cell morphology, viability) [1] [20] | Target-based assay (e.g., enzyme inhibition, binding) [1] |
| Key Challenge | Target deconvolution and identification [1] [16] | Developing physiologically relevant assays; compound cell permeability [16] |
| Typical Output | Novel drug target and a bioactive compound [1] | Validated phenotype linked to a known target [1] |
| Relation to Classical Terms | Analogous to "forward pharmacology" or "phenotypic drug discovery" (PDD) [17] | Analogous to "reverse pharmacology" or "target-based drug discovery" (TDD) [17] |
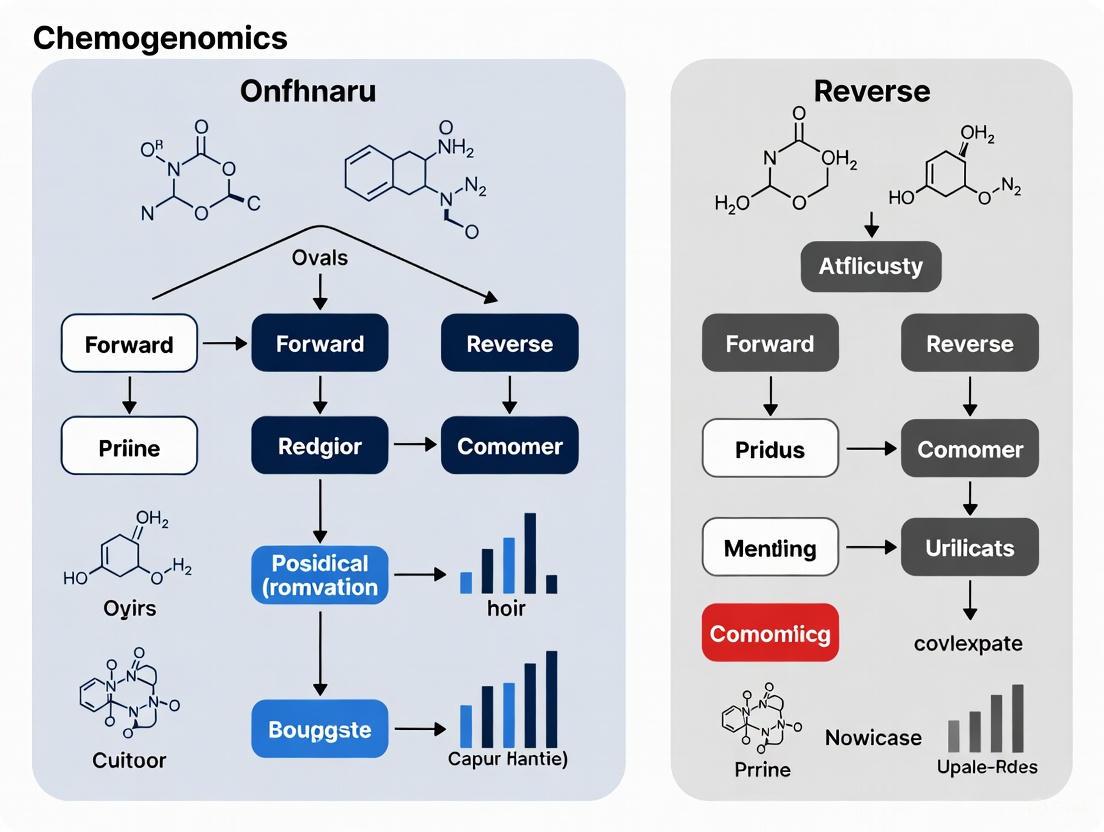
Diagram 1: Forward vs. Reverse Chemogenomics Workflows.
Core Concepts and Technologies
Phenotypic Screening
Phenotypic screening is a target-agnostic technique that tests compounds in biologically relevant model systems to identify those that cause a desirable change in phenotype, such as altered cell morphology, proliferation, or protein expression [20] [17].
Key Applications and Rationale: The strength of phenotypic screening is its ability to identify compounds that exert a therapeutic effect through novel, unanticipated mechanisms of action (MOA). A landmark analysis by Swinney and Anthony found that phenotypic screening was responsible for the discovery of a majority of first-in-class small-molecule drugs approved between 1999 and 2008 [18] [17]. This is largely because the cellular context inherently accounts for critical factors like cell permeability, metabolic stability, and complex pathway interactions, which are major causes of failure in drug development [16]. Phenotypic assays are particularly valuable when the disease-relevant target is unknown or cannot be easily isolated for a reductionist assay [20].
Technology and Data Handling: Modern phenotypic screening is synonymous with High-Content Screening (HCS). HCS utilizes automated microscopy and multiplexed fluorescent staining to simultaneously capture multiple phenotypic parameters from individual cells [20]. These systems, such as the Opera Phenix Plus, generate vast amounts of high-quality image data from 2D or 3D cell cultures [20]. The subsequent challenge is data management and analysis. Powerful image analysis platforms (e.g., Image Artist) are required to extract quantitative data on dozens of features, including cell shape, organelle distribution, and protein localization and intensity [20]. This multi-parametric data allows for a nuanced assessment of a compound's overall effect on the biological system.
Target-Based Assays
Target-based assays represent a hypothesis-driven approach. They begin with the selection of a specific molecular target (e.g., a kinase, receptor, or protease) hypothesized to play a critical role in a disease pathway. Compounds are then screened for their ability to modulate the activity of this purified target in vitro [16] [17].
Key Applications and Rationale: The primary advantage of target-based screening is its clarity of mechanism. From the outset, researchers know the intended target of a hit compound, which simplifies the subsequent optimization process [16]. This approach is highly amenable to High-Throughput Screening (HTS) of vast chemical libraries, often comprising millions of compounds, because the assays (e.g., fluorescence-based enzymatic assays) are typically homogenous and easy to automate [16] [11]. While phenotypic screening has an edge in discovering first-in-class drugs, target-based approaches have been highly productive for developing "best-in-class" drugs that improve upon the profile of a pioneer drug [16].
Limitations and Evolution: A significant limitation of traditional target-based assays is their reductionist nature. A compound that is potent against a purified protein may fail in a cellular environment due to poor permeability, off-target effects, or compensation within a biological network [16]. To address this, the field is increasingly adopting "targeted phenotypic" or "sweet spot" approaches. These are cell-based assays where the primary readout is the activity or localization of a specific, engineered target (e.g., phosphorylation of a downstream protein, translocation of a transcription factor), thus combining the mechanistic clarity of a target-based approach with the physiological context of a phenotypic assay [16].
Navigating Chemogenomic Space
Chemogenomics is the system-level strategy that connects chemical and biological space. It is founded on the principle that related targets (within a protein family) will interact with related compounds [1]. The "chemogenomic space" encompasses the intersection of all possible drug-like molecules with all potential drug targets in the genome [1].
Experimental Strategies: A common method is to create a targeted chemical library enriched with known ligands for several members of a protein family. Since ligands for one family member often show affinity for others, this library can be used to systematically probe the entire family, identifying ligands for previously "orphan" receptors and elucidating their function [1]. Experimentally, chemogenomics relies on profiling the response of every gene to a small molecule. A powerful example is the use of barcoded yeast deletion libraries (YKO collection). In these assays, pooled deletion strains are grown competitively in the presence of a drug. Monitoring the relative abundance of each strain's barcode via sequencing reveals which gene deletions make the cell sensitive or resistant to the compound, generating a fitness-based chemogenomic profile that offers profound insight into the drug's MOA [19].
Computational and AI-Driven Approaches: The scale of chemogenomic space makes it a prime application for computational methods. Computer-Aided Drug Discovery (CADD) and artificial intelligence (AI) are now used to model protein networks against large compound libraries, dramatically accelerating the exploration of this space [11]. Companies like Recursion and Exscientia use AI to analyze high-content phenotypic data (phenomics) and generative chemistry to design novel compounds, effectively creating a closed-loop design-make-test-analyze cycle [21]. Furthermore, chemogenomic profiling can be used for drug repositioning; by comparing the gene expression signature of a known drug to signatures of diseases or other drugs, new therapeutic indications can be identified [1] [11].
Table 2: Comparative Analysis of Screening Approaches
| Aspect | Phenotypic Screening | Target-Based Screening |
|---|---|---|
| Definition | Identifies compounds that alter cellular/organism phenotype without prior target knowledge [20] [17] | Identifies compounds that modulate a specific, predefined molecular target [16] [17] |
| Primary Readout | Multi-parametric cellular changes (morphology, growth, protein distribution) [20] | Specific target activity (e.g., enzyme inhibition, receptor binding) [16] |
| Throughput | Moderate (increasing with advanced HCS) [16] | High (amenable to ultra-HTS) [16] [11] |
| Key Advantage | Physiologically relevant, identifies novel mechanisms, accounts for permeability/toxicity [18] [20] | Clear mechanism of action, highly scalable, efficient for lead optimization [16] |
| Major Challenge | Target deconvolution is complex and time-consuming [16] [17] | May not translate to cellular/ in vivo context; can be biologically simplistic [16] |
| Success Bias | More successful for first-in-class medicines [18] [17] | More productive for best-in-class medicines [16] |
Experimental Protocols and Methodologies
Protocol 1: A Forward Chemogenomic Workflow Using a High-Content Phenotypic Screen
This protocol outlines a typical forward chemogenomic screen to identify compounds that reverse a disease-associated cellular phenotype, followed by target deconvolution.
1. Assay Development and Optimization:
- Cellular Model Selection: Choose a disease-relevant cell model. This could be a cancer cell line, primary patient-derived cells, or induced pluripotent stem cell (iPSC)-derived neurons. Increasingly, 3D spheroids or organoids are used for greater physiological relevance [16] [22].
- Phenotypic Readout: Define and validate a quantifiable, disease-relevant phenotypic endpoint. Examples include: synapse count in a neurodegenerative model, mitochondrial morphology in a metabolic disease model, or tumor cell invasion in a 3D matrix [20] [22].
- Assay Validation: Establish robust assay parameters, including Z'-factor (a statistical measure of assay quality), cell seeding density, and compound incubation time. Test known control compounds to ensure the assay can detect the desired phenotype reversal [22].
2. Primary Screening and Hit Stratification:
- Screening: Use a confocal high-content imaging system (e.g., Opera Phenix Plus) to screen a chemical library. The system automatically acquires images from multi-well plates and analyzes them using pre-configured algorithms to extract hundreds of morphological features per cell [20].
- Hit Identification: Compounds are ranked based on their ability to normalize the disease phenotype. A hit stratification process follows, which may involve using multi-parametric data to cluster hits based on their phenotypic "fingerprint," helping to group compounds with similar MOAs even before target identification [20] [22].
3. Target Deconvolution (The Forward Chemogenomics Challenge):
- Chemogenomic Profiling: A powerful method is to use a fitness-based chemogenomic profile. In yeast, this involves competing the entire pool of barcoded deletion strains against the hit compound. Strains whose genes are required for survival in the presence of the compound will drop out of the pool. Sequencing the barcodes identifies these sensitive strains, pointing to the potential pathway or even the direct target of the compound [19].
- Affinity Purification: Chemoproteomic approaches can be used. The hit compound is immobilized on a resin and used as bait to pull down interacting proteins from a cell lysate. The bound proteins are then identified via mass spectrometry [17].
- Transcriptional Profiling: The RNA expression profile of cells treated with the hit compound can be compared to a reference database of expression profiles from cells treated with compounds of known MOA. A "guilt-by-association" principle is applied: if the unknown compound's profile matches that of a known compound, it may share a similar target or pathway [19].
Protocol 2: A Reverse Chemogenomic Workflow for Kinase Inhibitor Profiling
This protocol starts with a target-based screen and progresses to phenotypic validation, a classic reverse chemogenomics strategy.
1. In Vitro Target-Based Screening:
- Target and Assay: Select a purified kinase protein (the target) and establish a homogenous, biochemical assay to measure its activity. A common format uses an ATP-consuming reaction coupled to a fluorescent or luminescent readout.
- High-Throughput Screening (HTS): Screen a diverse compound library or a kinase-focused targeted library. The assay is run in 1536-well plates to maximize throughput. Hits are identified based on a statistical threshold of inhibition (e.g., >70% inhibition at 10 µM) [1] [11].
2. Counter-Screening and Selectivity Profiling:
- Selectivity Assessment: To avoid non-specific inhibitors, counter-screen hits against a panel of related kinases and unrelated enzymes. This helps identify compounds with desirable selectivity profiles early.
- Cellular Target Engagement: Confirm that the compound engages the intended target in a cellular environment. Techniques include cellular thermal shift assays (CETSA) or using engineered reporter cell lines that signal when the pathway is modulated [16].
3. Phenotypic Validation in a Disease Model:
- Phenotypic Assay: Test the confirmed hit compounds in a disease-relevant cellular model. For a kinase inhibitor in oncology, this could be a cell proliferation assay or a 3D tumor spheroid invasion assay.
- High-Content Analysis: If the phenotypic endpoint is complex (e.g., differentiation state, cytoskeletal rearrangement), use high-content imaging to quantitatively measure the compound's effect, thereby linking the specific target inhibition to a systems-level phenotypic outcome [16]. This completes the reverse chemogenomics loop.
Diagram 2: Integrated Discovery Workflow Combining Forward and Reverse Approaches.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The effective execution of phenotypic, target-based, and chemogenomic studies relies on a suite of specialized reagents and tools.
Table 3: Key Research Reagent Solutions
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| High-Content Screening System (e.g., Opera Phenix Plus) [20] | Automated, high-resolution imaging for phenotypic screening. | Confocal imaging, simultaneous multi-channel acquisition, live-cell capability, water immersion lenses for 3D models. |
| Phenotypic Assay Microplates (e.g., PhenoPlate) [20] | Supports cell growth and imaging for HCS. | Optimal optical clarity, black walls to reduce crosstalk, tissue culture-treated surface, biologically inert. |
| Barcoded Yeast Deletion Collection (YKO) [19] | Genome-wide competitive fitness profiling for target deconvolution and MOA studies. | Pooled knockout strains, each with unique DNA barcodes, enables quantitative sequencing-based fitness measurement. |
| Targeted Chemical Libraries [1] | Focused compound sets for screening specific target families (e.g., kinases, GPCRs). | Enriched with known pharmacophores for the target family, increases hit rate for reverse chemogenomics. |
| AI/ML Drug Discovery Platform (e.g., Recursion OS, Exscientia's Centaur Chemist) [21] | Integrates and analyzes multi-omic and chemical data to design and prioritize compounds. | Uses generative AI for compound design, ML for analyzing HCS data, enables predictive in silico models. |
The historical dichotomy between phenotypic and target-based screening is giving way to a more integrated and synergistic paradigm. The evidence is clear: neither approach is superior in all contexts. Phenotypic screening's strength in identifying novel biology and target-based screening's efficiency in optimization are complementary forces in the modern drug discovery arsenal [16]. The framework of chemogenomic space, navigated through the parallel strategies of forward and reverse chemogenomics, provides a powerful conceptual map for this integration.
The future of the field is being shaped by several key trends. First, the adoption of more complex and physiologically relevant models—such as iPSCs, organoids, and microphysiological systems ("organs-on-chips")—is bridging the gap between traditional in vitro assays and human physiology, promising better translational outcomes [16] [22]. Second, the explosion of AI and machine learning is revolutionizing every step of the process. AI can now analyze high-dimensional phenotypic data to predict MOA, design novel compounds de novo, and even propose new therapeutic hypotheses from vast knowledge graphs [21] [7]. The recent merger of companies like Recursion (with its massive phenomic data) and Exscientia (with its generative chemistry AI) exemplifies the drive to create end-to-end, AI-powered discovery platforms [21].
Finally, the concept of the "informacophore" is emerging as a data-driven evolution of the traditional pharmacophore. It represents the minimal set of structural and machine-learned features essential for biological activity, identified through the analysis of ultra-large chemical datasets, thereby reducing reliance on biased human intuition [7]. As these technologies mature, the distinction between forward and reverse chemogenomics may blur, giving rise to a continuous, iterative discovery loop where phenotypic observations and target-level insights constantly inform one another, dramatically accelerating the journey from pattern to pill.
The Synergy with Genomics and Proteomics in Systematic Screening
Abstract Systematic screening represents a paradigm shift in biomedical research, moving from a reductionist focus on single targets to a global, integrative approach for identifying novel therapeutic targets and bioactive compounds. This methodology is fundamentally powered by the synergy between genomics and proteomics, which provide complementary layers of biological information. Within the strategic framework of chemogenomics, systematic screening bifurcates into two powerful, complementary approaches: forward chemogenomics, which begins with a phenotypic screen in a biological system to identify active compounds before target deconvolution, and reverse chemogenomics, which starts with a defined molecular target to screen for modulating compounds. This whitepaper provides an in-depth technical guide to the experimental protocols, data types, and bioinformatics tools that underpin this integrated strategy, offering a roadmap for researchers and drug development professionals to leverage these technologies for accelerated discovery.
Functional genomics and proteomics constitute a global, systematic, and comprehensive approach to identifying the processes and pathways involved in both normal and diseased physiological states [23]. Systematic screening in this context involves the parallel interrogation of thousands of biological molecules—be they transcripts or proteins—to decipher the complex mechanisms underlying disease and treatment responses. The ultimate aim of this integrative genomics approach is to understand pathophysiological processes, identify genes/proteins suitable for diagnostics, and discover novel therapeutic targets [23].
The high-throughput nature of these technologies generates immense, complex datasets, necessitating powerful bioinformatics tools for data processing, quality control, and interpretation. The integration of multi-omics data through systematic screening is thus transforming cancer treatment and personalized medicine, facilitating the discovery of biomarkers and the development of individualized therapeutic plans [24].
Chemogenomics: The Strategic Framework
Chemogenomics is an emerging discipline that combines the latest tools of genomics and chemistry, applying them to target and drug discovery. It aims to eliminate the bottleneck in target identification by measuring the broad, conditional effects of chemical libraries on whole biological systems or by efficiently screening large chemical libraries against selected targets [25]. This field operates on two primary axes:
2.1 Forward Chemogenomics In forward chemogenomics, active compounds are identified based on their conditional phenotypic effect on a whole biological system (e.g., a cell line or model organism) rather than on their inhibition of a specific protein target. This "phenotype-first" approach is followed by the subsequent study of the mechanistic basis of the observed phenotype, a process known as target deconvolution [25]. This strategy is particularly valuable for identifying novel biological pathways and mechanisms without preconceived notions about the specific proteins involved.
2.2 Reverse Chemogenomics Reverse chemogenomics begins with gene sequences of interest that are expressed as target proteins and screened in a high-throughput, target-based manner against compound libraries. This approach places particular emphasis on the parallel exploration of gene and protein families based on the structure–activity relationship homology concept [25]. It represents a more targeted, hypothesis-driven approach to drug discovery.
Genomic and Proteomic Data Types in Systematic Screening
Table 1: Core Omics Data Types in Systematic Screening
| Data Type | Technology Examples | Measured Elements | Application in Screening |
|---|---|---|---|
| Genomics | Whole-Genome Sequencing (WGS), Whole-Exome Sequencing (WES) [24] | SNPs, Copy Number Variations, Structural Variants [24] | Identify genetic alterations associated with disease susceptibility and treatment response. |
| Transcriptomics | RNA Sequencing (RNA-seq), DNA Microarrays [23] [24] | Gene Expression Levels, Transcript Isoforms, Gene Fusions [24] | Uncover expression signatures and pathway activities in response to compounds or in diseased states. |
| Proteomics | 2D Gel Electrophoresis, Mass Spectrometry [23] | Protein Expression, Post-Translational Modifications (e.g., Phosphorylation) [23] | Detect functional effectors, protein isoforms, and activation states not evident from genomic data. |
The human proteome is significantly more complex than the genome, with an estimated one million human proteins, far exceeding the number of genes, due to mechanisms like alternative splicing and post-translational modifications [23]. This complexity makes the integration of proteomic data with genomic information particularly critical for a complete understanding of biological systems.
Experimental Protocols for Integrated Screening
4.1 Protocol for a Forward Chemogenomics Workflow
Objective: To identify compounds inducing a desired phenotype (e.g., cell death in a specific cancer cell line) and subsequently identify their molecular targets.
Phenotypic Screening:
- Cell Culture: Maintain disease-relevant cell lines (e.g., cancer cells with specific genetic background) under standard conditions.
- Compound Library Treatment: Treat cells with a diverse chemical library. Include positive and negative controls on each screening plate.
- Phenotypic Assay: After a defined incubation period, measure the phenotypic endpoint (e.g., cell viability using ATP-based assays, apoptosis via caspase activation, or image-based morphometric analysis).
- Hit Selection: Identify "hit" compounds that produce a statistically significant effect on the phenotype beyond a predefined threshold (e.g., >50% inhibition of viability).
Target Deconvolution via Genomics and Proteomics:
- Transcriptomic Profiling: Treat the cell line with the hit compound and perform RNA-seq [24] to obtain genome-wide expression profiles. Compare to untreated controls to identify differentially expressed genes and pathways.
- Proteomic Profiling: In parallel, lyse the treated cells. Separate proteins using two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), which resolves proteins by their isoelectric point (pI) in the first dimension and molecular weight in the second [23]. Excise protein spots that are differentially expressed and identify them using mass spectrometry (e.g., MALDI-TOF/TOF) [23].
- Bioinformatic Integration: Integrate the transcriptomic and proteomic data using pathway analysis tools (e.g., Ingenuity Pathway Analysis, GSEA) [24] to pinpoint consistently altered biological pathways and generate hypotheses about the primary molecular target.
Target Validation: Validate the putative target using techniques such as CRISPR knockout, RNAi knockdown, or cellular thermal shift assays (CETSA) to confirm that the phenotypic effect is dependent on or correlated with target engagement.
4.2 Protocol for a Reverse Chemogenomics Workflow
Objective: To discover compounds that modulate the activity of a predefined, high-value target (e.g., a kinase implicated in cancer).
Target Selection and Production:
- Selection: Select a target protein based on genetic evidence (e.g., mutations in TCGA database [24]) or pathway analysis.
- Expression and Purification: Clone the gene of interest into an expression vector. Express and purify the recombinant protein from a suitable system (e.g., E. coli, insect cells).
High-Throughput Target-Based Screening:
- Assay Development: Develop a robust biochemical assay to measure the target's activity (e.g., kinase activity measured via ATP consumption or substrate phosphorylation).
- Primary Screening: Screen the compound library against the purified target. Use high-density miniaturized assays (e.g., in 1536-well plates) to increase throughput [25].
- Hit Confirmation: Re-test primary hits in dose-response curves to determine potency (IC50/EC50) and exclude false positives.
Functional Validation in Cellular Context:
- Cellular Assays: Test confirmed hits in cell-based assays to verify target engagement and functional effects (e.g., inhibition of downstream pathway phosphorylation).
- Multi-Omic Profiling: Treat responsive cell models with the lead compound and perform integrated genomic (RNA-seq) and proteomic (mass spectrometry) analyses to characterize the system-wide effects of target inhibition and identify potential mechanism-based toxicity or resistance pathways [24]. This step is critical for understanding the broader context of target modulation.
Essential Research Reagents and Tools
Table 2: Key Research Reagent Solutions for Integrated Screening
| Reagent / Tool Category | Specific Examples | Function / Explanation |
|---|---|---|
| Omics Databases | The Cancer Genome Atlas (TCGA), Genomic Data Commons (GDC) [24] | Provide large-scale, publicly available genomic, transcriptomic, and clinical datasets for target prioritization and validation. |
| Bioinformatics Tools | ANNOVAR, cBioPortal, Ingenuity Pathway Analysis (IPA), GSEA [24] | Used for variant annotation, visualization of cancer genomics data, and pathway/network analysis of omics data. |
| Compound Libraries | Annotated Compound Libraries, Designed Libraries [25] | Collections of chemical compounds with known bioactivity or designed around specific protein families, used for screening. |
| Sequencing Platforms | Illumina (SBS), PacBio (SMRT), Oxford Nanopore [24] | Enable WGS, WES, and RNA-seq for comprehensive genomic and transcriptomic profiling. |
| Proteomics Platforms | 2D Gel Electrophoresis, Mass Spectrometry (e.g., MALDI-TOF/TOF) [23] | Separate and identify proteins, including post-translational modifications, from complex biological samples. |
Visualization of Workflows and Relationships
The following diagrams, created using Graphviz DOT language, illustrate the core workflows and conceptual relationships described in this guide.
The synergy between genomics and proteomics provides an unparalleled, multi-dimensional view of biological systems, making systematic screening a cornerstone of modern drug discovery. The complementary strategies of forward and reverse chemogenomics offer powerful, flexible frameworks for tackling this complexity, enabling the simultaneous identification of therapeutic targets and bioactive compounds. As high-throughput technologies continue to evolve and bioinformatics tools become more sophisticated, the integration of these omics data streams will undoubtedly yield novel biomarkers, deeper mechanistic insights, and more effective, personalized therapeutic strategies for complex diseases like cancer.
Execution and Impact: Practical Workflows and Real-World Applications
Forward chemogenomics represents a paradigm shift in modern drug discovery, moving away from target-centric approaches toward an unbiased, biology-first methodology. This approach systematically screens chemical libraries against cellular or organismal models to identify compounds that induce a specific, desired phenotype without presupposing a molecular target [3] [1]. The core premise is that by starting with a biologically relevant outcome—such as inhibition of cancer cell growth or reduction of a pathological marker—researchers can work backward from the phenotypic hit to identify therapeutically relevant drug targets that might otherwise remain undiscovered [6] [1]. This methodology has gained significant momentum with advancements in high-content screening, functional genomics, and artificial intelligence, enabling the capture of subtle, disease-relevant phenotypes at unprecedented scale and resolution [3].
The strategic positioning of forward chemogenomics within the broader chemogenomics landscape distinguishes it fundamentally from its reverse counterpart. Forward chemogenomics begins with a phenotypic screen to find molecules that produce a specific biological effect, subsequently identifying the protein targets responsible [6] [1]. Conversely, reverse chemogenomics starts with a specific, known protein target and screens for molecules that interact with it, later validating the phenotypic effects—an approach more aligned with traditional target-based drug discovery [1]. This distinction is critical; forward chemogenomics is ideally suited for exploring complex biological systems where the key molecular players are unknown, while reverse chemogenomics excels when a validated target requires ligand optimization [1].
Table 1: Core Strategic Differences Between Forward and Reverse Chemogenomics
| Feature | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotype of interest (e.g., cell death, differentiation) | Known protein target (e.g., kinase, GPCR) |
| Screening Focus | Phenotypic changes in cells or organisms | Binding or functional modulation of a specific protein |
| Primary Goal | Identify novel drug targets and their modulators | Find ligands for a predefined target |
| Key Challenge | Deconvoluting the molecular target of active compounds | Demonstrating phenotypic relevance of target engagement |
| Ideal Application | Complex diseases with poorly understood etiology | Well-validated target families with known biology |
The Experimental Workflow: From Phenotype to Target
The implementation of a forward chemogenomics campaign requires a meticulously planned, multi-stage workflow. Each stage builds upon the last, transforming a macroscopic biological observation into a validated, druggable target.
Phenotypic Assay Design and Compound Screening
The initial and most critical phase involves designing a robust, biologically relevant phenotypic assay. The assay must accurately capture a disease-relevant process and be amenable to high- or medium-throughput screening [3] [1]. For example, an assay for an anticancer phenotype might measure the inhibition of tumor cell invasion in a three-dimensional matrix, while a neuroprotective phenotype could assess neuronal survival under oxidative stress. Key to success is designing phenotypic assays that can lead directly to target identification, which remains a significant challenge in the field [1].
Advanced technologies now enable highly compressed and information-rich phenotypic screens. Methods such as Pooled Perturb-seq allow for the compressed screening of multiple genetic or chemical perturbations simultaneously, with computational deconvolution dramatically reducing sample size, labor, and cost while maintaining data richness [3]. High-content imaging, often using assays like Cell Painting, provides a powerful way to visualize multiple cellular components and generate rich morphological profiles that serve as a detailed fingerprint of a compound's activity [3].
Target Deconvolution and Validation
Once compound(s) producing the desired phenotype are confirmed, the challenging phase of target deconvolution begins—identifying the specific protein(s) responsible for the observed effect.
Several experimental methodologies are employed for target deconvolution:
- Affinity Purification: The active compound is immobilized on a solid support and used as bait to capture binding proteins from cell lysates. Identified proteins are then validated through functional studies.
- Genome-Wide Profiling: Techniques like genome-wide CRISPR knockout or RNAi screens can identify genes whose loss modifies the compound's phenotypic effect, pointing to potential targets or pathway components.
- Transcriptomic/Proteomic Profiling: Comparing the gene expression or protein abundance signatures of active compounds to databases of signatures for compounds with known mechanisms can suggest a shared molecular target [3].
- Chemogenomic Libraries: Using targeted libraries enriched for compounds known to interact with specific protein families (e.g., kinases, GPCRs) can provide immediate clues about the target class involved [6] [1].
A compelling example of successful target deconvolution comes from the application of machine learning-based approaches like idTRAX, which has been used to identify cancer-selective targets in triple-negative breast cancer by integrating multiple data layers from phenotypic screens [3].
Diagram 1: Forward Chemogenomics Workflow
The Scientist's Toolkit: Essential Research Reagents and Platforms
The successful execution of a forward chemogenomics campaign relies on a suite of specialized reagents, tools, and platforms. These resources enable the generation of high-quality, interpretable data at each stage of the process.
Table 2: Essential Research Reagents and Platforms for Forward Chemogenomics
| Tool/Reagent | Function and Application | Examples / Key Features |
|---|---|---|
| Targeted Chemical Libraries | Pre-selected compound collections focused on specific protein families; provide initial target hypotheses. | Pfizer Chemogenomic Library, GSK Kinase Inhibitor Set, LOPAC1280 [6] |
| Cell Painting Assay | High-content imaging assay using fluorescent dyes to visualize multiple organelles; generates rich morphological profiles. | Uses dyes for nucleus, ER, mitochondria, Golgi, actin, cytoplasm [3] |
| Perturb-seq | Single-cell RNA sequencing of cells under genetic or chemical perturbation; links phenotype to transcriptome. | Genome-scale Perturb-seq captures subtle, disease-relevant phenotypes [3] |
| AI/ML Integration Platforms | AI-powered platforms that integrate multimodal data (imaging, omics) to identify patterns and predict MoA. | Ardigen's PhenAID, Archetype AI, idTRAX [3] |
| CRISPR Knockout Libraries | Genome-wide or focused gene knockout pools for functional genomic screens to identify target genes. | Used in modifier screens to identify genes that affect compound sensitivity [1] |
Case Studies in Forward Chemogenomics
Application in Oncology and Infectious Disease
Forward chemogenomics has yielded significant successes in identifying novel therapeutic strategies in complex disease areas. In oncology, the Archetype AI platform was used with patient-derived phenotypic data to identify AMG900 and novel invasion inhibitors for lung cancer, demonstrating how computational backtracking of phenotypic shifts can reveal viable drug candidates without initial target knowledge [3]. Similarly, during the COVID-19 pandemic, the DeepCE model predicted gene expression changes induced by novel chemicals, enabling high-throughput phenotypic screening for antiviral compounds. This integrative approach generated new lead compounds consistent with clinical evidence, showcasing the power of combining phenotypic and omics data with AI for rapid drug repurposing [3].
Mode of Action Elucidation for Traditional Medicines
A particularly innovative application of forward chemogenomics is in elucidating the molecular mechanisms underlying traditional medicines. For Traditional Chinese Medicine (TCM) and Ayurveda, where the precise mode of action is often unknown, chemogenomic approaches have been used to predict ligand targets relevant to known phenotypes [1]. For a class of TCM known as "toning and replenishing medicine," computational target prediction identified sodium-glucose transport proteins and PTP1B as targets linked to the hypoglycemic phenotype, providing a molecular rationale for the traditional use [1].
Challenges and Future Perspectives
Despite its promise, forward chemogenomics faces several practical challenges. Data heterogeneity and sparsity from different formats, ontologies, and resolutions complicate integration, and many datasets are too sparse for effective training of advanced AI models [3]. Target deconvolution remains inherently difficult, often requiring multiple, orthogonal approaches to confidently identify the protein responsible for a phenotype [1]. Furthermore, issues of data privacy, model interpretability, and the need for substantial computational infrastructure present ongoing hurdles [3].
The future of forward chemogenomics is inextricably linked to advances in artificial intelligence and multi-omics integration. AI/ML models are increasingly capable of fusing multimodal datasets—including electronic health records, high-content imaging, multi-omics, and sensor data—into unified models that enhance predictive performance [3]. As these technologies mature, the integration of phenotypic screening with omics and AI will evolve from a specialized approach into a new operating system for drug discovery, enabling the systematic identification of novel drug targets and therapeutic strategies for complex diseases.
Chemogenomics represents a systematic approach in modern drug discovery that investigates the interactions between targeted chemical libraries and families of functionally related proteins. The core premise of chemogenomics is that focused chemical libraries can be screened against protein families to identify novel ligands and simultaneously elucidate the functions of uncharacterized proteins [1]. This approach has emerged as a powerful alternative to traditional one-target-one-drug discovery methods, particularly for complex diseases involving multiple molecular pathways [26]. Within this paradigm, two complementary strategies have emerged: forward chemogenomics and reverse chemogenomics.
Forward chemogenomics (phenotype-based) begins with screening compounds against a desired cellular phenotype to identify active molecules, followed by target deconvolution to identify the macromolecular partners responsible for the observed effect [1]. This approach is analogous to classical forward genetics but uses chemical perturbagens instead of genetic mutations. In contrast, reverse chemogenomics (target-based) starts with a specific protein target of interest and screens focused chemical libraries to identify modulators, then characterizes the resulting phenotypic effects to validate the target's functional role in a biological context [27] [1]. This review focuses specifically on the methodology, applications, and implementation of reverse chemogenomics as a systematic approach for target validation and drug discovery.
Conceptual Framework: Reverse Chemogenomics Workflow
The reverse chemogenomics approach operates on the principle that small molecules can serve as precise tools to establish causal relationships between protein targets and phenotypic outcomes. As illustrated in Figure 1, this methodology involves a sequential process from target selection to phenotypic validation.
Figure 1: Reverse Chemogenomics Workflow
The process begins with target identification, where a specific protein target is selected based on genomic, proteomic, or bioinformatic evidence suggesting its potential role in a disease pathway [27] [28]. Next, focused library screening involves testing a targeted chemical library against the selected protein target using high-throughput or virtual screening methods [1] [26]. The hit compounds identified through this process are then advanced to phenotypic characterization in cellular or organismal models to determine the biological consequences of target modulation [27] [1]. Finally, the target validation step establishes whether the protein target is indeed physiologically relevant to the disease process, based on the concordance between chemical modulation and phenotypic outcome [27].
This systematic approach allows researchers to move from a hypothetical target to validated biology using chemical tools as mechanistic probes. The strength of reverse chemogenomics lies in its ability to provide direct causal evidence linking specific protein targets to phenotypic changes, bridging the gap between in vitro biochemistry and complex biological systems.
Computational Methodologies for Reverse Screening
A critical technical component of reverse chemogenomics is the computational prediction of compound-target interactions, known as reverse screening or target fishing. These methods identify potential protein targets for a given compound by screening against databases of known targets, serving as a efficient starting point for experimental validation [13]. Three primary computational approaches have been developed, each with distinct principles and applications as summarized in Table 1.
Table 1: Computational Reverse Screening Methods for Target Identification
| Method | Basic Principle | Representative Tools | Required Data | Key Advantages |
|---|---|---|---|---|
| Shape Screening | Compares 3D molecular shape and electrostatic properties | ChemMapper, TargetHunter | Ligand database with target annotations | Scaffold independence; handles conformational flexibility |
| Pharmacophore Screening | Matches essential chemical features for biological activity | PharmMapper, Schrödinger Phase | Pharmacophore database with target annotations | Identifies key interaction points; less sensitive to scaffold differences |
| Reverse Docking | Docks compound into binding sites of multiple protein structures | INVDOCK, idTarget | Protein 3D structure database | Provides structural binding mode; estimates binding affinity |
Shape Screening Methods
Shape screening operates on the principle that compounds with similar three-dimensional shapes may bind to the same protein targets, even if they possess different chemical scaffolds [13]. This approach involves comparing the overall molecular shape and electrostatic properties of a query compound against a database of known ligands with annotated targets. When a query molecule demonstrates high shape similarity to a database ligand, the targets of that ligand become candidate targets for the query molecule [13].
Key tools in this category include ChemMapper, which utilizes molecular access system (MACCS) fingerprints for 2D similarity comparisons or 3D shape-based alignment, and TargetHunter, which employs extended-connectivity fingerprints (ECFP4 and ECFP6) for structural similarity searching [13]. Shape screening is particularly valuable for identifying novel scaffold hops, where chemically distinct compounds interact with the same biological target through complementary spatial arrangements.
Pharmacophore Screening Methods
Pharmacophore screening extends beyond molecular shape to identify essential chemical features required for biological activity, such as hydrogen bond donors/acceptors, hydrophobic regions, aromatic rings, and charged groups [13]. This method involves creating a pharmacophore model - an abstract representation of molecular interactions - from a query compound and screening it against a database of pharmacophore models derived from known ligands or protein binding sites.
PharmMapper is a prominent publicly available server that uses pharmacophore matching to identify potential targets from a large collection of pharmacophore models derived from protein-ligand complexes in the Protein Data Bank [13]. Commercial packages like Schrödinger's Phase also offer comprehensive pharmacophore-based screening capabilities. This approach is particularly effective when the query compound shares limited structural similarity with known ligands but contains critical functional groups that can engage similar interaction motifs in protein binding sites.
Reverse Docking Methods
Reverse docking represents the most computationally intensive approach, involving the systematic docking of a query compound into a collection of protein binding sites to identify potential interactions based on complementary steric and energetic factors [13]. Unlike conventional docking that seeks ligands for a single target, reverse docking screens one compound against multiple targets successively.
Tools such as INVDOCK (one of the earliest reverse docking programs) and idTarget employ algorithms to score and rank potential protein targets based on predicted binding affinities or complementary surface matching [13]. The success of reverse docking depends critically on the quality and diversity of the protein structure database, with common sources including the Protein Data Bank (PDB), sc-PDB (a database of druggable binding sites), and other curated collections of protein structures with relevant binding sites.
Experimental Implementation and Protocols
Building Focused Chemical Libraries
The foundation of successful reverse chemogenomics lies in the construction of appropriate chemical libraries tailored to the target family of interest. A well-designed focused library contains compounds that collectively sample the chemical space likely to interact with members of a specific protein family while providing sufficient diversity to identify selective probes [26].
Library Design Principles: Targeted chemical libraries for reverse chemogenomics typically include known ligands for at least some members of the protein family, capitalizing on the principle that ligands designed for one family member often show affinity for other related proteins [1]. For example, a kinase-focused library would include ATP-competitive compounds with scaffolds known to interact with the conserved kinase domain, while also incorporating allosteric inhibitors and structurally diverse compounds to maximize the probability of identifying hits against both characterized and orphan kinases [26].
Library Size and Composition: Practical focused libraries for experimental screening typically contain 1,000-10,000 compounds, balancing comprehensiveness with practical screening constraints [26]. The NIH Molecular Libraries Program, for instance, established a collection of ~300,000 compounds for broader screening, but targeted reverse chemogenomics efforts often employ more focused sets [15]. These libraries should include both known drugs (for repurposing opportunities) and specialized tool compounds with optimized pharmacological properties [26].
Table 2: Essential Research Reagents for Reverse Chemogenomics
| Reagent Category | Specific Examples | Function in Reverse Chemogenomics |
|---|---|---|
| Focused Chemical Libraries | Pfizer chemogenomic library, GSK Biologically Diverse Compound Set, NCATS MIPE library | Provide targeted compound sets for specific protein families with known target annotations |
| Target Protein Resources | Recombinant proteins, cell lines expressing target proteins, tissue samples | Enable in vitro and cellular screening assays against targets of interest |
| Screening Platforms | High-throughput screening systems, high-content imaging (Cell Painting) | Facilitate rapid compound screening and multiparametric phenotypic assessment |
| Bioactivity Databases | ChEMBL, PubChem, BindingDB, ExCAPE-DB | Supply annotated compound-target interaction data for computational predictions |
| Target Validation Tools | siRNA/shRNA libraries, CRISPR-Cas9 systems, antibody arrays | Enable orthogonal validation of target engagement and functional relevance |
Experimental Protocol: Target-Based Screening Followed by Phenotypic Validation
The following protocol outlines a standard reverse chemogenomics approach for target validation:
Step 1: Target Selection and Assay Development
- Select a protein target based on genomic, proteomic, or bioinformatic evidence of disease relevance [28]
- Develop a robust biochemical or biophysical assay to measure compound-target engagement
- For enzymes: implement activity assays (e.g., fluorescence-based, luminescence)
- For receptors: develop binding assays (e.g., SPR, FRET, radioligand displacement)
- Validate assay performance (Z' factor >0.5, signal-to-background ratio >3:1)
Step 2: Primary Screening
- Screen the focused chemical library against the target using the developed assay
- Employ appropriate controls (positive, negative, DMSO controls)
- Use concentration typically between 1-10 μM for initial single-point screening
- Define hit criteria (e.g., >50% inhibition/activation compared to controls)
Step 3: Hit Confirmation and Selectivity Profiling
- Confirm primary hits in dose-response format (typically 8-12 point curves)
- Determine potency metrics (IC50, EC50, Ki)
- Assess selectivity against related targets (e.g., same protein family)
- Exclude promiscuous binders/aggregators using counter-screens (e.g., detergent sensitivity)
Step 4: Phenotypic Characterization
- Test confirmed hits in cellular models relevant to the disease context
- Assess functional consequences of target engagement (e.g., pathway modulation, phenotypic changes)
- Establish correlation between target modulation and phenotypic effect
- Determine cellular potency (EC50) and maximal response
Step 5: Target Validation
- Use orthogonal approaches to confirm target engagement in cells (e.g., CETSA, CRISPR)
- Demonstrate phenotype reversal with genetic approaches (e.g., RNAi, knockout)
- Establish dose-response and temporal relationship between target engagement and phenotype
- Confirm specificity using structurally distinct chemotypes targeting the same protein
Case Study: Application to Peptidoglycan Biosynthesis Targets
A practical example of reverse chemogenomics comes from antibacterial discovery, where researchers applied a ligand library originally developed for the murD enzyme (involved in peptidoglycan synthesis) to other members of the mur ligase family (murC, murE, murF, murA, and murG) [1]. Through chemogenomics similarity principles, known murD ligands were mapped to other mur ligases to identify new targets for existing compounds. Structural studies and molecular docking revealed candidate ligands for murC and murE ligases, demonstrating how reverse chemogenomics can expand the target spectrum of existing chemical tools and identify potential broad-spectrum antibacterial agents [1].
Effective reverse chemogenomics relies on integrating diverse data sources to build comprehensive compound-target interaction networks. Key resources include:
Chemogenomics Databases: Large-scale databases such as ExCAPE-DB integrate over 70 million structure-activity relationship data points from public sources (PubChem and ChEMBL), providing standardized compound structures, target information, and activity annotations [29]. These resources enable predictive modeling of polypharmacology and off-target effects by providing comprehensive coverage of chemical and target spaces.
Target Annotation Resources: Databases like ChEMBL (containing ~1.7 million molecules with bioactivities against ~11,000 unique targets) and DrugBank (integrating drug data with target information) provide critical annotations linking compounds to their protein targets [26] [29]. These resources are essential for building targeted chemical libraries and interpreting screening results.
Network Pharmacology Platforms: Systems like the one developed by [26] integrate ChEMBL, KEGG pathways, Gene Ontology, Disease Ontology, and morphological profiling data from Cell Painting assays in a graph database (Neo4j). This enables the connection of compound-target interactions with pathway context and phenotypic outcomes, facilitating the interpretation of reverse chemogenomics screening data within broader biological networks.
Reverse chemogenomics has established itself as a powerful systematic approach for target validation, leveraging focused chemical libraries to establish causal relationships between proteins and phenotypes. By combining computational prediction of compound-target interactions with experimental validation, this approach provides a robust framework for elucidating protein function and identifying therapeutic opportunities.
The continued growth of chemogenomics databases, improved computational methods for target prediction, and the development of more sophisticated focused libraries will further enhance the power of reverse chemogenomics. Integration with functional genomics data, such as from CRISPR screens, and advances in structural biology will provide additional layers of evidence for target validation [15] [30]. Furthermore, the application of machine learning to chemogenomics data holds promise for predicting novel compound-target interactions beyond what is possible with similarity-based methods alone [28] [29].
As these technologies mature, reverse chemogenomics will play an increasingly important role in bridging the gap between genomic discoveries and therapeutic applications, particularly for rare diseases and neglected conditions where traditional drug discovery approaches have proven challenging. The systematic framework provided by reverse chemogenomics offers a path forward for validating novel therapeutic targets and expanding the druggable genome.
The drug discovery paradigm has significantly shifted from a reductionist "one target–one drug" vision to a more complex systems pharmacology perspective that acknowledges a single drug often interacts with several targets [26]. This evolution has been propelled by the high attrition rates of drug candidates in advanced clinical stages due to lack of efficacy or safety, particularly for complex diseases like cancer and neurological disorders which frequently arise from multiple molecular abnormalities [26]. Within this context, chemogenomics has emerged as a systematic approach to screening targeted chemical libraries against specific drug target families with the dual goals of identifying novel drugs and elucidating the functions of less-characterized targets [1].
Chemogenomics operates through two complementary experimental frameworks. Forward chemogenomics investigates a particular phenotype of interest to identify small molecules that induce this phenotype, subsequently using these modulators as tools to pinpoint the responsible proteins [1]. This approach aligns with classical phenotypic drug discovery. Conversely, reverse chemogenomics begins by identifying small compounds that perturb a specific enzyme's function in vitro, then analyzes the phenotypic consequences induced by these molecules in cellular or whole-organism systems [1]. This strategy mirrors traditional target-based approaches but enhanced by parallel screening capabilities across target families. The strategic application of both frameworks relies critically on two foundational elements: well-designed phenotypic assays capable of detecting relevant biological changes, and precisely constructed compound libraries that maximize the potential for identifying bioactive molecules.
Core Concepts: Compound Categories and Library Composition
Classification of Bioactive Compounds
Compounds deployed in biological screening can be systematically categorized into three distinct classes—tools, probes, and drugs—each with defined characteristics and applications [31].
Tool compounds are broadly applied to understand general biological mechanisms, often serving essential roles in cell biology research. Examples include cycloheximide, used to study translational mechanisms, and forskolin, which stimulates adenylate cyclase and serves as a critical tool for developing assays for Gαi/Gαs coupled GPCRs [31]. While some tools like doxycycline function in both basic research and therapeutic contexts, others such as cycloheximide are considered too toxic for in vivo applications but remain invaluable for in vitro studies [31].
Chemical probes are specifically designed to modulate isolated target proteins or signaling pathways with high potency and selectivity [31]. Optimal chemical probes exhibit well-defined structure-activity relationships (SARs) where both active and inactive analogs are identified, along with favorable properties regarding stability, solubility, and cell permeability [31]. Notable examples include PD0325901, a selective allosteric MEK1/2 inhibitor used to probe this kinase both in vitro and in vivo, and UNC0638, a lysine methyltransferase inhibitor that enables exploration of this enzyme's function in model systems [31].
Drugs represent the most recognized category of small molecules, distinguished by their proven pharmacological benefits in clinical settings [31]. However, drugs constitute the exception in small molecule research due to stringent requirements for bioavailability, low toxicity, and metabolic stability [31]. Some drugs with specific targets, such as Sildenafil (phosphodiesterase inhibitor), can function effectively as chemical probes, while others with undefined or complex mechanisms of action may be unsuitable for probing specific biological pathways [31].
Table 1: Characteristics of Compound Categories in Screening Libraries
| Category | Primary Application | Key Properties | Examples |
|---|---|---|---|
| Tool Compounds | Understanding general biological mechanisms | May have toxicity limitations for in vivo use; widely applied to in vitro assays | Cycloheximide, Forskolin, Actinomycin D, Doxycycline |
| Chemical Probes | Modulating isolated targets or pathways | High potency, selectivity, established SAR, favorable physicochemical properties | PD0325901 (MEK1/2 inhibitor), UNC0638 (lysine methyltransferase inhibitor), K-trap (HDAC inhibitor) |
| Drugs | Therapeutic applications | Optimized ADME properties, clinical safety and efficacy established | Sildenafil, Fludarabine phosphate, Bambuterol, Ethacrynic acid |
Historical Evolution of Library Design
The composition of chemical libraries has evolved significantly, reflecting accumulating biological knowledge and changing discovery paradigms. Originally, compound collections from companies such as Ciba Geigy and Bayer emerged from the dye industry, with successful repurposing of dyes leading to the first chemotherapeutics [31]. The chance discovery of chlordiazepoxide (Librium) from quinazolone-3-oxides represented an early example of leveraging privileged scaffolds [31]. The concept of "privileged structures"—chemical scaffolds with high bioactivity across multiple receptor types—was formally recognized in 1988, establishing a rationale for biology-oriented library design [31].
Modern libraries typically incorporate historical archives, compounds from drug discovery programs (including related analogs and clinical candidates), and commercial sources encompassing both purified natural products and combinatorial collections [31]. Contemporary strategies include natural product-inspired libraries and diversity-oriented synthesis to explore new regions of chemical property space [31]. The accumulation of extensive bioassay data on compound libraries has created rich databases that provide an archaeological footprint of past discovery efforts, enabling more informed library design strategies [31].
Designing Phenotypic Assays for Chemogenomics
Advanced Phenotypic Profiling Technologies
Modern phenotypic screening leverages sophisticated technologies including induced pluripotent stem (iPS) cell technologies, gene-editing tools such as CRISPR-Cas, and advanced imaging assays [26]. Among these, high-content imaging-based high-throughput phenotypic profiling has emerged as a particularly powerful approach. The "Cell Painting" assay represents a prominent example, utilizing multiple fluorescent dyes to reveal cellular components followed by automated image analysis to extract morphological features [26].
In a typical Cell Painting implementation, U2OS osteosarcoma cells are plated in multiwell plates, perturbed with test treatments, stained, fixed, and imaged on a high-throughput microscope [26]. Automated image analysis using software like CellProfiler identifies individual cells and measures hundreds of morphological features across different cellular compartments (cell, cytoplasm, and nucleus) [26]. These parameters can include intensity, size, area shape, texture, entropy, correlation, granularity, and spatial relationships [26]. The resulting morphological profiles enable researchers to group compounds into functional pathways, identify phenotypic impacts of chemical perturbations, and discover signatures of disease [26].
Diagram 1: High-content phenotypic screening workflow
Quantitative High-Throughput Screening (qHTS) Implementation
Quantitative high-throughput screening (qHTS) represents a significant advancement over traditional HTS by performing multiple-concentration experiments in low-volume cellular systems using high-sensitivity detectors [32]. This approach screens large chemical libraries across a range of concentrations, offering lower false-positive and false-negative rates compared to single-concentration screening [32]. Modern implementations, such as those in the US Tox21 collaboration, can simultaneously test more than 10,000 chemicals across 15 concentrations [32].
The Hill equation (HEQN) remains the most common nonlinear model for describing qHTS concentration-response relationships [32]. The logistic form of the HEQN is:
[ Ri = E0 + \frac{(E\infty - E0)}{1 + \exp{-h[\log Ci - \log AC{50}]}} ]
Where (Ri) represents the measured response at concentration (Ci), (E0) is the baseline response, (E\infty) is the maximal response, (AC{50}) is the concentration for half-maximal response, and (h) is the shape parameter [32]. The (AC{50}) and (E{max}) (calculated as (E\infty - E_0)) parameters are frequently used to approximate compound potency and efficacy, respectively [32].
Critical Considerations in qHTS Data Analysis
Parameter estimation from qHTS data presents significant statistical challenges, particularly when using the Hill equation model [32]. Estimates can be highly variable if the tested concentration range fails to include at least one of the two HEQN asymptotes, responses are heteroscedastic, or concentration spacing is suboptimal [32]. Research has demonstrated that (AC_{50}) estimates show poor repeatability when the concentration range does not adequately define the response curve, with estimates sometimes spanning several orders of magnitude [32].
Table 2: Impact of Experimental Design on Parameter Estimation in qHTS
| Experimental Condition | Effect on AC50 Estimation | Impact on Emax Estimation | Recommended Approach |
|---|---|---|---|
| Both asymptotes defined (AC50 = 0.1 μM, Emax ≥50%) | Precise estimation (narrow confidence intervals) | Reliable estimation | Ideal scenario - use standard HEQN fitting |
| Only lower asymptote defined (AC50 = 10 μM, Emax = 100%) | Precise estimation | Reliable estimation | Suitable for HEQN fitting |
| Incomplete asymptote definition (AC50 = 0.001 μM, Emax = 25%) | Poor repeatability (wide confidence intervals spanning orders of magnitude) | Unreliable estimation | Use alternative approaches or improve concentration range |
| Increased replication (n=3 or n=5 per concentration) | Noticeable improvement in precision across all conditions | Moderate improvement in precision | Implement when feasible within screening constraints |
| Non-monotonic response profiles | HEQN fundamentally unsuitable | HEQN fundamentally unsuitable | Employ non-parametric or alternative modeling approaches |
Several strategies can enhance qHTS reliability. Including experimental replicates improves measurement precision, with larger sample sizes leading to noticeable increases in the precision of both (AC{50}) and (E{max}) estimates [32]. However, researchers must remain cognizant of potential systematic errors introduced by factors such as well location effects, compound degradation, signal bleaching, or compound carryover between plates [32]. Additionally, not all substances exhibit sigmoidal concentration-response relationships within tested ranges, necessitating complementary approaches with reliable classification performance across diverse profile types [32].
Constructing Targeted Compound Libraries
Design Principles for Chemogenomic Libraries
Effective chemogenomic library design requires strategic consideration of multiple factors, including library size, cellular activity, chemical diversity, availability, and target selectivity [33]. A common method involves including known ligands for at least one—and preferably several—members of the target family, leveraging the principle that ligands designed for one family member often bind to additional relatives [1]. This approach ensures the collective compounds in a targeted library should bind to a high percentage of the target family [1].
Recent work has demonstrated the feasibility of designing minimal screening libraries with extensive target coverage. For precision oncology applications, researchers have developed a minimal screening library of 1,211 compounds targeting 1,386 anticancer proteins, demonstrating that well-designed compact libraries can maintain broad target coverage [33]. Such libraries can be further refined through scaffold analysis—a method that decomposes molecules into representative core structures through systematic removal of terminal side chains and stepwise ring reduction to identify characteristic core structures [26].
Integration with Network Pharmacology
Modern chemogenomic library design increasingly incorporates network pharmacology approaches that integrate heterogeneous data sources to model complex drug-target-pathway-disease relationships [26]. This involves combining bioactivity data (from sources like ChEMBL), pathway information (from KEGG, Gene Ontology), disease ontologies, and morphological profiling data within graph databases such as Neo4j [26]. This integrative framework enables identification of proteins modulated by chemicals that correlate with morphological perturbations at the cellular level, potentially leading to identifiable phenotypes or disease associations [26].
Diagram 2: Strategic framework for targeted compound library design
Implementation in Precision Medicine
Targeted compound libraries have demonstrated particular utility in precision oncology applications. In a pilot screening study targeting glioblastoma (GBM), researchers utilized a physical library of 789 compounds covering 1,320 anticancer targets to profile glioma stem cells from patients [33]. The resulting cell survival profiling revealed highly heterogeneous phenotypic responses across patients and GBM subtypes, highlighting the potential of targeted libraries to identify patient-specific vulnerabilities [33]. This approach exemplifies the reverse chemogenomics strategy, where compounds with known targets are used to characterize phenotypic responses in disease-relevant cellular models.
Research Reagent Solutions: Essential Materials for Chemogenomics Studies
Table 3: Essential Research Reagents for Advanced Chemogenomics Studies
| Reagent Category | Specific Examples | Function in Chemogenomics Research |
|---|---|---|
| Chemical Libraries | Pfizer chemogenomic library, GSK Biologically Diverse Compound Set (BDCS), Prestwick Chemical Library, Sigma-Aldrich Library of Pharmacologically Active Compounds, NCATS MIPE library [26] | Provide curated collections of bioactive compounds for screening; foundation for both phenotypic and target-based approaches |
| Cell-Based Assay Systems | U2OS osteosarcoma cells (for Cell Painting), induced pluripotent stem cells (iPSCs), patient-derived primary cells [26] [33] | Serve as biological systems for phenotypic screening; patient-derived cells enable personalized therapeutic approaches |
| Staining Reagents | Cell Painting dye set (multiple fluorescent dyes targeting different cellular compartments) [26] | Enable multiplexed morphological profiling by revealing cellular components through high-content imaging |
| Bioactivity Databases | ChEMBL database, KEGG pathways, Gene Ontology, Human Disease Ontology [26] | Provide annotated bioactivity, target, pathway, and disease relationship data for network pharmacology approaches |
| Analysis Software | CellProfiler (image analysis), ScaffoldHunter (scaffold analysis), Neo4j (graph database), R packages (clusterProfiler, DOSE, ggplot2) [26] | Enable morphological feature extraction, chemical scaffold analysis, network integration, and statistical analysis of screening data |
The synergistic application of well-designed phenotypic assays and targeted compound libraries creates powerful workflows for both forward and reverse chemogenomics approaches. In forward chemogenomics, a phenotypic assay (such as Cell Painting or another disease-relevant cellular model) serves as the starting point for identifying compounds that induce a desired phenotype, followed by target deconvolution using the annotated compounds in the library [1] [26]. Conversely, in reverse chemogenomics, compounds with known target annotations from the library are applied to phenotypic assays to validate their biological effects and potentially discover new therapeutic applications [1].
The integration of advanced phenotypic profiling with richly annotated chemical libraries and network pharmacology frameworks represents a powerful paradigm for modern drug discovery. This approach moves beyond single-target thinking to embrace the complexity of biological systems, accelerating the identification of novel therapeutic agents and their mechanisms of action. As these technologies continue to evolve, they promise to enhance both the efficiency and success rates of drug discovery, particularly for complex diseases that have proven resistant to traditional single-target approaches.
The systematic investigation of Traditional Medicine (TM), including Traditional Chinese Medicine (TCM) and Ayurveda, represents a significant frontier in modern drug discovery. These ancient medical systems provide extensive natural resources for medicinal compounds, generally regarded as effective and safe based on centuries of human use [34]. However, the complexity of multi-compound formulations and their multi-target mechanisms presents substantial challenges for scientific validation using conventional pharmacological approaches [34]. This case study explores how modern chemogenomics—the systematic screening of targeted chemical libraries against families of related drug targets—provides powerful methodological frameworks for elucidating these complex mechanisms [1].
The fundamental challenge in TM research lies in dissecting the molecular mechanisms of herbal medicines at a holistic level. TCM formulations, or "Fangji," are designed under the principle of "syndrome differentiation" with obvious multiple-compound characteristics, creating complex interactions among biological systems, drugs, and complex diseases [34]. Within chemogenomics, two complementary paradigms exist: forward chemogenomics (phenotype-based) and reverse chemogenomics (target-based). This case study examines how both approaches facilitate the deconvolution of TM mechanisms, highlighting specific applications, experimental protocols, and significant findings that bridge traditional knowledge with modern scientific validation.
Chemogenomics Framework: Forward vs. Reverse Approaches
Chemogenomics integrates target and drug discovery by using active compounds as probes to characterize proteome functions [1]. The interaction between a small compound and a protein induces a phenotype that, when characterized, enables researchers to associate proteins with molecular events [1]. Compared with genetic approaches, chemogenomics techniques modify the function of a protein rather than the gene and allow observation of interactions and reversibility in real-time [1].
Table 1: Fundamental Comparison of Chemogenomics Approaches
| Feature | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Desired phenotype in cells or organisms | Known compound or protein target |
| Primary Objective | Identify compounds inducing phenotype, then find targets | Find compounds binding specific target, then validate phenotype |
| Screening Approach | Phenotypic screening | Target-based screening |
| Typical Assays | Cell-based phenotypic assays, whole-organism models | In vitro enzymatic tests, binding assays |
| Key Challenge | Designing assays that lead directly from screening to target identification | Parallel screening and lead optimization across target families |
| Application in TM | Identifying active components in complex mixtures based on biological activity | Validating suspected molecular targets of traditional formulations |
Forward Chemogenomics Strategy
In forward chemogenomics (also called classical chemogenomics), researchers study a particular phenotype and identify small compounds that interact with this function while the molecular basis remains unknown [1]. Once modulators are identified, they serve as tools to identify the proteins responsible for the phenotype [1]. For example, a loss-of-function phenotype could represent arrested tumor growth. Once compounds leading to this target phenotype are identified, the subsequent step involves identifying the corresponding gene and protein targets [1]. The National Cancer Institute's NCI60 screen exemplifies this approach, where anti-proliferative effects of compounds on cancer cell lines are recorded to differentiate classes of anti-proliferative agents and generate mechanistic hypotheses [35].
Reverse Chemogenomics Strategy
In reverse chemogenomics, researchers first identify small compounds that perturb the function of an enzyme in the context of an in vitro enzymatic test [1]. After identifying modulators, they analyze the molecule-induced phenotype in cellular tests or whole organisms [1]. This method helps confirm the role of the enzyme in the biological response [1]. This strategy resembles traditional target-based approaches but is enhanced by parallel screening and the ability to perform lead optimization on multiple targets belonging to one target family [1]. Reverse chemogenomics often employs techniques like reverse screening or "target fishing" to identify protein targets for known active compounds [13].
Figure 1: Workflow comparison of forward and reverse chemogenomics approaches for traditional medicine research.
Case Study: TCM 'Toning and Replenishing Medicine'
Background and Phenotypic Evidence
The 'toning and replenishing medicine' class in TCM has demonstrated various therapeutic phenotypes in experimental models, including anti-inflammatory, antioxidant, neuroprotective, hypoglycemic activity, immunomodulatory, antimetastatic, and hypotensive effects [1] [36]. Despite these well-documented phenotypic responses, the molecular mechanisms underlying these diverse effects remained largely uncharacterized until the application of chemogenomics approaches.
Forward Chemogenomics Analysis
Researchers initially applied forward chemogenomics principles by studying the hypoglycemic phenotype observed with certain TCM formulations [36]. Using phenotypic screening approaches, they identified bioactive compounds that produced glucose-lowering effects in cellular and animal models. The subsequent target identification phase represented the critical challenge in this workflow. By employing chemogenomics profiling and in silico target prediction tools, researchers hypothesized that sodium-glucose transport proteins (SGLT1 and SGLT2) and protein tyrosine phosphatase 1B (PTP1B)—an insulin signaling regulator—served as potential molecular targets relevant to the observed hypoglycemic activity [1] [36]. This hypothesis was further supported by established biological knowledge: SGLT transporters play crucial roles in glucose absorption, while PTP1B functions as a key negative regulator of insulin signaling [36].
Reverse Chemogenomics Validation
The reverse chemogenomics approach complemented these findings by starting with the chemical structures of compounds present in the TCM formulations. Researchers performed reverse screening—a computational method that identifies potential protein targets for a given compound by screening against databases of known target-ligand interactions or protein structures [13]. Through this approach, they confirmed that compounds from 'tonifying and replenishing medicinal' classes exhibited binding potential to SGLT1, SGLT2, and PTP1B [36]. This reverse chemogenomics validation strengthened the mechanistic hypothesis generated through forward approaches, creating a convergent understanding of the molecular mechanisms underlying the traditional formulations' efficacy.
Table 2: Experimentally Validated Targets for TCM Toning Formulations
| Target Protein | Biological Function | Predicted Activity | Experimental Validation |
|---|---|---|---|
| SGLT1 | Intestinal glucose absorption | Hypoglycemic | Glucose uptake assays [36] |
| SGLT2 | Renal glucose reabsorption | Hypoglycemic | Transport inhibition studies [36] |
| PTP1B | Insulin signaling regulation | Insulin sensitizer | Enzyme inhibition assays [36] |
| GPBAR1 | Metabolic regulation | Metabolic modulation | Receptor activation studies [36] |
Experimental Protocols for Chemogenomics in TM
Protocol 1: Forward Chemogenomics Workflow for Phenotype-Driven Target Discovery
Objective: Identify molecular targets of TM formulations with observed phenotypic effects but unknown mechanisms.
Materials and Reagents:
- Traditional medicine extract or purified compounds
- Relevant cell lines or model organisms
- Phenotypic assay reagents (cell viability, glucose uptake, etc.)
- Compound libraries for screening
- Genomic or proteomic profiling tools
Procedure:
- Phenotypic Screening: Establish robust assays quantifying the phenotypic effect of interest (e.g., glucose uptake assay for hypoglycemic activity) [36].
- Bioactive Compound Identification: Fractionate TM extracts and test fractions in phenotypic assays to identify active components [34].
- Chemogenomic Profiling: Treat model systems with active compounds and generate genome-wide response profiles (transcriptomic, proteomic, or fitness profiles) [19].
- Reference Comparison: Compare chemogenomic profiles against reference databases of genetic perturbations or compound treatments with known mechanisms [19].
- Target Hypothesis Generation: Identify candidate targets based on profile similarities to reference perturbations affecting specific pathways [19].
- Experimental Validation: Validate candidate targets through direct binding assays, genetic perturbation studies, or biochemical assays [1].
Protocol 2: Reverse Screening for Target Fishing of TM Compounds
Objective: Identify protein targets for characterized TM compounds using computational and experimental approaches.
Materials and Reagents:
- Pure TM compounds
- Target protein libraries or databases
- Computational resources for virtual screening
- Binding assay reagents (SPR, thermal shift, etc.)
- Cell culture materials for phenotypic confirmation
Procedure:
- Compound Preparation: Obtain 3D structures of TM compounds through energy minimization and conformational analysis [13].
- Database Curation: Compile high-quality protein target databases with annotated binding sites (e.g., PDB, ChEMBL, BindingDB) [29] [13].
- Shape Screening: Compare 3D molecular shapes or 2D fingerprints against annotated ligand databases to identify similar compounds with known targets [13].
- Pharmacophore Screening: Match essential chemical features of query molecules against pharmacophore models of protein binding sites [13].
- Reverse Docking: Perform molecular docking of TM compounds against multiple protein targets to identify favorable interactions [13].
- Consensus Prediction: Integrate results from multiple reverse screening methods to generate high-confidence target predictions [13].
- Experimental Confirmation: Validate computational predictions through direct binding assays and functional studies in biological systems [13].
Figure 2: Integrated reverse screening workflow for target identification of traditional medicine compounds.
Research Reagents and Computational Tools
Successful implementation of chemogenomics approaches for TM research requires specialized reagents, databases, and computational tools. The table below summarizes essential resources for establishing a TM chemogenomics platform.
Table 3: Essential Research Reagents and Resources for TM Chemogenomics
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Chemical Databases | ChEMBL, PubChem, BindingDB | Source of annotated compound-target interactions for reverse screening [29] [13] |
| Target Databases | Protein Data Bank (PDB), ExCAPE-DB | Repository of protein structures and chemogenomics data for target fishing [29] [13] |
| Computational Tools | PharmMapper, INVDOCK, ChemMapper | Software for pharmacophore screening, reverse docking, and shape similarity calculations [13] |
| Chemogenomics Libraries | LOPAC1280, Prestwick Library, NIH Molecular Libraries Program | Curated compound collections with known bioactivities for reference profiling [6] |
| Bioactivity Profiling | Yeast Knockout (YKO) Collection, Gene Expression Profiling | Tools for genome-wide chemogenomic response measurement [19] |
| Structure Standardization | AMBIT, Chemistry Development Kit | Software for chemical structure curation and standardization prior to screening [29] |
Data Integration and Validation Frameworks
Analytical Considerations for TM Studies
The complex nature of TM formulations presents unique challenges for chemogenomics studies. Unlike single-compound therapeutics, TM typically contains multiple bioactive components that may act synergistically on multiple targets. Researchers must employ specific analytical strategies to address these challenges:
- Network Pharmacology Integration: Construct compound-target-pathway networks to visualize and analyze multi-target mechanisms [34].
- Synergistic Effect Analysis: Design experiments to distinguish between additive, synergistic, or antagonistic effects of compound combinations [34].
- Polypharmacology Assessment: Evaluate the potential for single compounds to interact with multiple targets, which may contribute to overall efficacy [6].
- ADME Filtering: Apply absorption, distribution, metabolism, and excretion (ADME) criteria to prioritize biologically relevant compounds from complex mixtures [34].
Validation Strategies
Rigorous validation remains essential for establishing credible mechanisms of action for TM interventions:
- Direct Binding Assays: Confirm predicted compound-target interactions using surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), or thermal shift assays [13].
- Functional Cellular Assays: Demonstrate that target engagement translates to relevant phenotypic changes in disease-relevant cell models [1].
- Genetic Validation: Use gene knockdown, knockout, or overexpression to confirm target contribution to the observed phenotype [19].
- Disease Model Correlation: Validate mechanistic findings in animal models that recapitulate aspects of human disease [36].
Forward and reverse chemogenomics provide complementary frameworks for bridging the gap between traditional knowledge and modern mechanistic understanding in traditional medicine. Forward chemogenomics offers a phenotype-driven approach that identifies bioactive components and their molecular targets without predetermined assumptions about mechanism. Conversely, reverse chemogenomics provides target-focused strategies that efficiently map established compounds to their protein targets and biological pathways. Together, these approaches enable the systematic deconvolution of complex traditional medicine formulations into well-defined compound-target interactions, validated mechanisms of action, and ultimately, novel therapeutic opportunities grounded in both traditional wisdom and contemporary science. As chemogenomics methodologies continue to advance—particularly through improvements in computational prediction, data integration, and experimental validation—they promise to accelerate the discovery of biologically active compounds from traditional medicine sources while providing mechanistic insights that support their rational application in modern therapeutic contexts.
This technical guide provides an in-depth examination of chemogenomics strategies for targeting pharmaceutically relevant gene families, with a focused analysis on kinases and G protein-coupled receptors (GPCRs). The content is structured within the conceptual framework of forward versus reverse chemogenomics approaches, detailing experimental protocols, data analysis methods, and visualization techniques essential for researchers and drug development professionals. By integrating computational predictions with high-throughput experimental validation, chemogenomics enables systematic exploration of chemical space against biological target families, accelerating the identification of novel therapeutic agents and their mechanisms of action.
Chemogenomics represents a systematic framework for screening targeted chemical libraries against specific drug target families such as GPCRs, nuclear receptors, kinases, and proteases with the ultimate goal of identifying novel drugs and drug targets [1]. This approach integrates target and drug discovery by using active compounds as probes to characterize proteome functions, generating specific phenotypes through compound-protein interactions that can be systematically analyzed [6]. The fundamental principle underpinning chemogenomics is that related targets within a gene family often share structural similarities in their binding sites, meaning that ligands designed for one family member may also interact with other members of the same family [1].
The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention, and chemogenomics strives to study the intersection of all possible drugs on all these potential targets [1]. This paradigm represents a generalization of traditional Quantitative Structure-Activity Relationship (QSAR) methods; whereas QSAR predicts interactions for a single protein, chemogenomic models can concurrently predict interactions for multiple proteins across chemical space [6]. The strategy is particularly suitable for targets with some known ligands, enabling the identification of ligands for important therapeutic target groups including enzymes, GPCRs, and ion channels [6].
Table 1: Key Characteristics of Chemogenomics Approaches
| Feature | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotypic observation | Target protein |
| Primary Goal | Identify drug targets | Validate phenotypes |
| Screening Method | Phenotypic assays on cells/animals | Target-based in vitro assays |
| Typical Applications | Functional characterization of orphan targets, mode of action studies | Lead optimization, selectivity profiling |
| Key Challenge | Target deconvolution | Relevance of in vitro findings to physiology |
Chemogenomics Framework: Forward vs. Reverse
The experimental practice of chemogenomics is broadly categorized into two complementary approaches: forward (classical) chemogenomics and reverse chemogenomics. These strategies differ in their starting points and objectives but share the common goal of connecting chemical compounds with biological targets and functions.
Forward Chemogenomics
In forward chemogenomics, researchers begin with a particular phenotype of interest and identify small compounds that interact with this function, even when the molecular basis of the phenotype is unknown [1]. Once modulators are identified, they serve as tools to identify the proteins responsible for the phenotype. For example, in a loss-of-function scenario such as arrest of tumor growth, compounds inducing this target phenotype are first identified, followed by identification of the corresponding gene and protein targets [1]. The main challenge in forward chemogenomics lies in designing phenotypic assays that efficiently lead from screening to target identification, requiring sophisticated target deconvolution strategies [6].
This approach has proven valuable in determining the mode of action for traditional medicines, where compounds with known phenotypic effects but unknown molecular targets are investigated. For instance, chemogenomics has been applied to identify mechanisms of action for Traditional Chinese Medicine and Ayurveda by predicting ligand targets relevant to known phenotypes [1]. In these cases, databases containing chemical structures alongside phenotypic effects enable in silico analysis to link traditional medicines with potential molecular targets.
Reverse Chemogenomics
Reverse chemogenomics begins with a known protein target and identifies small compounds that perturb its function in the context of an in vitro enzymatic test [1]. Once modulators are identified, the phenotype induced by the molecule is analyzed in cellular or whole organism contexts to confirm the biological role of the target. This approach essentially mirrors traditional target-based drug discovery but enhances it through parallel screening capabilities and the ability to perform lead optimization across multiple targets within the same family [6].
Reverse chemogenomics has been successfully applied to identify new antibacterial agents by leveraging existing ligand libraries for known bacterial enzymes. In one documented approach, researchers capitalized on the similarity principle within the mur ligase family, mapping known murD ligands to other family members (murC, murE, murF, murA, and murG) to identify new targets for existing ligands [1]. This strategy efficiently identified broad-spectrum Gram-negative inhibitors since the targeted peptidoglycan synthesis pathway is exclusive to bacteria.
Target Family Focus: GPCRs and Kinases
GPCR Chemogenomics
G protein-coupled receptors represent the largest family of membrane proteins and are targeted by approximately one-third of all FDA-approved drugs [37]. Their significance in physiology and therapeutics makes them prime candidates for chemogenomics approaches. GPCRs mediate vital biological functions by translating extracellular stimuli into intracellular actions through conformational changes that facilitate coupling to heterotrimeric G proteins and arrestins [37].
The chemogenomics approach to GPCR drug discovery has been revolutionized by advances in structural biology, with over 30 GPCR structures now determined [38]. These structural insights enable more rational design of targeted chemical libraries and improve computational prediction of ligand-receptor interactions. A key development in GPCR chemogenomics is the design of genome-wide pan-GPCR drug discovery platforms that systematically explore relationships between traditional medicines and the entire GPCRome [39]. These platforms employ uniform approaches to establish GPCR-expressing cell lines and examine connections between chemical compounds and GPCR families comprehensively.
GPCR ligands identified through chemogenomics approaches include various small molecules and peptides with diverse chemical structures, including alkaloids, flavonoids, furanochromones, glycosides, steroidal glycosides, and terpenoids [39]. Among these, alkaloids represent the most significant category, with at least 11 FDA-approved GPCR-targeting drugs being alkaloids, such as morphine from Papaver somniferum which targets opioid receptors [39].
Figure 1: GPCR Canonical Signaling Pathway - This diagram illustrates the fundamental GPCR signaling mechanism where extracellular ligand binding triggers intracellular G protein activation and downstream effector pathways.
Kinase Chemogenomics
While the search results provided limited specific information on kinase chemogenomics, the general principles of chemogenomics apply similarly to kinase target families. Kinases represent another pharmaceutically important gene family characterized by structural conservation in their ATP-binding pockets, making them particularly amenable to chemogenomics approaches. Targeted chemical libraries for kinases typically include ATP-mimetic compounds designed to interact with the conserved catalytic domain while achieving selectivity through interactions with unique subpockets and regions outside the active site.
The protein kinase inhibitor set from GlaxoSmithKline exemplifies a targeted chemogenomics library, comprising over 250 kinase-focused chemical probes that have been distributed to numerous collaborators in open-source research initiatives [6]. Such libraries enable systematic profiling of compound activity across multiple kinase family members, generating rich datasets that inform on selectivity patterns and structure-activity relationships.
Experimental Protocols and Methodologies
In Silico Chemogenomics Workflow
The computational workflow in chemogenomics integrates cheminformatics and bioinformatics approaches to predict drug-target interactions. The process begins with collection of protein structures and sequences for the gene family of interest, obtained from sources such as crystal structures, NMR data, biological homology models, or mutation data [6]. Molecules with known affinity profiles are compiled and used to train machine learning models that can predict activities for additional family members.
A key mathematical framework in chemogenomics involves representing target (t) and chemical (c) pairs by a vector Φ(t, c) to calculate a linear function f(t, c) = w⊤Φ(t, c), whose sign predicts binding potential between chemical c and target t [6]. Machine learning algorithms then calculate the vector w from training data about interacting and non-interacting pairs, enabling prediction of novel interactions. Deep learning approaches extend this framework through chemogenomic neural networks that take input from molecular graphs and protein sequence encoders to learn optimal combinations of molecule and protein representations [6].
Table 2: Key Experimental Assays in GPCR Chemogenomics
| Assay Type | Detection Method | Information Obtained | Throughput |
|---|---|---|---|
| Competitive Ligand Binding | Radioligand displacement or scintillation proximity | Direct binding affinity and kinetics | Medium |
| GTPγS Binding | Radioactive GTP analog | G protein activation | Medium |
| Second Messenger (cAMP, Ca2+) | Luminescence, fluorescence | Downstream signaling activation | High |
| β-arrestin Recruitment | Enzyme complementation (Presto-Tango) | G protein-independent signaling | High |
| BRET/FRET Biosensors | Energy transfer | Conformational changes and proximity | Medium |
High-Throughput Screening Platforms
High-throughput screening represents the experimental cornerstone of chemogenomics, enabling rapid evaluation of thousands to millions of compounds against target families [39]. For GPCR targets, screening techniques have evolved from traditional radioligand binding assays to sophisticated functional assays that detect various aspects of receptor activation and signaling.
The competitive ligand-binding assay remains a widely used method characterized by high specificity and sensitivity [39]. This technique quantifies interactions between GPCRs and radiolabeled ligands through titration with test molecules. Alternative nonradioactive assays have emerged to overcome limitations associated with radioisotopes, including fluorescence-based and luminescence-based detection methods.
For functional characterization, platforms like the GloSensor cAMP biosensor utilize a modified form of firefly luciferase containing a cAMP-binding motif to detect Gαs or Gαi-mediated signaling through luminescence readouts [40]. The Presto-Tango assay system measures β-arrestin recruitment by coupling GPCR C-termini to transcription factor activation, resulting in luminescence signals that reflect receptor internalization and G protein-independent signaling [40].
Figure 2: Chemogenomics Experimental Workflow - This diagram outlines the key stages in a comprehensive chemogenomics screening campaign, from target selection through hit validation.
The Scientist's Toolkit: Research Reagents and Materials
Successful implementation of chemogenomics approaches requires carefully selected research reagents and specialized materials. The following table details essential components for establishing chemogenomics screening platforms, particularly focused on GPCR and kinase target families.
Table 3: Essential Research Reagents for Chemogenomics Studies
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Targeted Chemical Libraries | Compound collections enriched for specific gene families | Protein Kinase Inhibitor Set (GSK), LOPAC1280, Pfizer Chemogenomic Library |
| Engineered Cell Lines | Recombinant cells expressing specific targets | GPCR-expressing cells with reporter genes (cAMP, β-arrestin) |
| Detection Reagents | Signal readout in various assay formats | GloSensor cAMP reagent, Europium-labeled ligands, fluorescent dye conjugates |
| Structural Biology Tools | Protein engineering for structural studies | T4 lysozyme fusions, BRIL fusion proteins, thermostabilizing mutations |
| Computational Resources | In silico prediction and modeling | Homology modeling software, molecular docking platforms, QSAR tools |
Data Analysis and Visualization
Analysis of chemogenomics data presents unique challenges due to the multidimensional nature of compound profiling across multiple targets. Activity landscape visualization methods have been developed to represent high-dimensional bioactivity spaces in intuitive formats that facilitate pattern recognition and hypothesis generation [41]. Network representations are particularly valuable for visualizing relationships between compounds and targets, highlighting clusters of activity and selectivity patterns.
For GPCR targets, detailed analysis of signaling bias requires comparison of compound activity across multiple assay formats measuring different signaling pathways. The relative activity of each agonist in one assay must be compared to its relative activity in other assays using the same reference agonist to yield a relative activity ratio that corrects for system bias and observational bias [40]. This rigorous analytical approach enables detection of true ligand bias between signaling pathways, which is crucial for developing therapeutics with improved efficacy and reduced side effects.
Advanced visualization techniques integrate structural information with functional data, mapping binding sites and residue interactions to understand the structural basis of selectivity and bias. For example, analysis of intracellular biased allosteric modulators has revealed how ligands engaging intracellular binding sites can promote pathway-biased signaling in cooperation with orthosteric ligands [40]. These visualizations help researchers understand how to design compounds with precise pharmacological properties.
Chemogenomics provides a powerful systematic framework for targeting gene families like kinases and GPCRs through integrated application of forward and reverse approaches. By combining computational prediction with experimental validation across target families, this paradigm accelerates the identification of novel therapeutic agents and their mechanisms of action. The continuing advances in structural biology, screening technologies, and data analysis methods promise to enhance the efficiency and effectiveness of chemogenomics strategies, enabling more comprehensive exploration of the intersection between chemical space and biological target space. As these methodologies mature, they will increasingly inform drug discovery pipelines and contribute to the development of safer, more effective therapeutics with precise mechanisms of action.
Navigating Challenges and Enhancing Success with AI and Data
Common Pitfalls in Forward Chemogenomics and Target Deconvolution Strategies
The drug discovery paradigm is fundamentally divided into two contrasting approaches: forward and reverse chemogenomics. In reverse chemogenomics (often termed target-based drug discovery), research begins with a known, validated molecular target. Scientists then aim to identify or design chemical compounds that interact with this specific target, subsequently testing for a desired phenotypic outcome [42]. Conversely, forward chemogenomics (phenotypic drug discovery) starts with a biological phenotype of interest. Compounds are screened for their ability to induce this phenotype, such as cell death or differentiation, after which the molecular targets responsible for the effect must be identified through target deconvolution [43] [42].
While forward chemogenomics offers the advantage of discovering first-in-class drugs with novel mechanisms of action, its primary challenge lies in the target deconvolution phase. This process is complex, resource-intensive, and fraught with potential missteps that can derail a drug development program [43]. This guide details the common pitfalls in these strategies and provides methodologies to overcome them, framing the discussion within the broader comparison of forward and reverse chemogenomics research.
Core Pitfalls in Target Deconvolution and Validation
The journey from a bioactive compound to a validated molecular target is fraught with technical and strategic challenges. These pitfalls can be categorized into computational, experimental, and validation-related issues.
Computational Target Prediction Pitfalls
Computational methods, which often provide the first hypothesis for a compound's target, face significant hurdles.
- Overreliance on Chemical Similarity: Many algorithms, including similarity ensemble approach (SEA) and PharmMapper, operate on the principle that chemically similar compounds share biological targets [43]. This can miss true targets for compounds with novel scaffolds and generate false positives for promiscuous chemotypes.
- Limited Proteome Coverage: Most in silico prediction tools are trained on known drug-target interactions, creating a bias toward well-characterized protein families like kinases and GPCRs. This leaves large portions of the "undrugged" proteome, including novel or uncharacterized targets, poorly covered [43].
- Neglect of Polypharmacology: Many computational models are designed to predict a single, primary target. However, drugs frequently interact with multiple proteins; on average, a drug binds between 6 and 12 different protein targets, and this polypharmacology can be central to its therapeutic efficacy [43]. Models that fail to account for this complexity provide an incomplete picture.
- The Translational Gap: A significant limitation of computational predictions is their frequent failure to translate to biological systems. For example, early in the COVID-19 pandemic, computational models predicted several drug repurposing candidates, such as hydroxychloroquine and lopinavir, which later showed no clinical benefit in randomized trials [44].
Experimental and Technical Pitfalls
Experimental strategies for target deconvolution, while powerful, come with their own set of technical limitations.
- Probe-Dependent Artifacts: Affinity-based pulldown and photoaffinity labeling (PAL) require modifying the compound of interest with a tag or handle (e.g., biotin, a photoreactive moiety) [42]. This chemical modification can alter the compound's cell permeability, binding affinity, or specificity, leading to a distorted view of its native interactions.
- Membrane Protein Challenges: Integral membrane proteins, such as GPCRs and ion channels, are notoriously difficult to study with standard affinity purification methods due to their hydrophobicity and reliance on the native lipid membrane for correct folding [42]. This is a critical omission as GPCRs alone are targets for approximately one-third of all marketed drugs [39].
- Focus on the Proteome: The vast majority of target deconvolution methods are designed to identify protein targets. This overlooks potential interactions with non-protein targets such as RNA, DNA, lipids, or metal ions, which can be critical for a drug's mechanism of action [43]. For instance, the antifungal drug amphotericin B functions by binding to the lipid ergosterol [43].
- Identification Without Functional Validation: Standard proteomics yields a list of putative protein binders, but it does not confirm which interactions are functionally relevant to the observed phenotype. Distinguishing causative on-target effects from incidental off-target binding requires extensive downstream validation [43].
Pitfalls in Target Prioritization and Validation
Once a list of putative targets is generated, the process of prioritization and validation introduces further risks.
- The "Most Significant" Fallacy: It is tempting to prioritize targets based solely on statistical significance from proteomic data (e.g., lowest p-value or highest abundance in pulldown). However, the most statistically significant hit is not always the most biologically relevant to the therapeutic phenotype [43].
- Insufficient Orthogonal Validation: Relying on a single method for validation is a common but critical error. Scrupulous validation requires multiple orthogonal methods to establish both direct binding of the compound to the target and a functional link to the phenotype in a relevant cellular context [43].
- Ignoring Polypharmacology in Validation: Using single-gene knockout or knockdown to validate a target can be misleading if the compound's efficacy relies on simultaneous modulation of multiple targets. If only one target is knocked out, the compound may still produce a phenotype through its other targets, leading to a false negative conclusion about the knocked-out target's importance [43].
The table below summarizes these key pitfalls and the associated risks for drug discovery projects.
Table 1: Common Pitfalls and Associated Risks in Target Deconvolution
| Category | Specific Pitfall | Impact on Research |
|---|---|---|
| Computational | Overreliance on chemical similarity | Missed novel targets; false positives for promiscuous chemotypes |
| Computational | Limited proteome coverage | Bias towards well-characterized protein families (e.g., kinases) |
| Computational | Neglect of polypharmacology | Incomplete understanding of a compound's full mechanism of action |
| Experimental | Probe-dependent artifacts (e.g., affinity tags) | Altered compound behavior; non-native interactions |
| Experimental | Difficulty with membrane proteins | Overlooks critical target classes like GPCRs and ion channels |
| Experimental | Focus solely on protein targets | Misses RNA, DNA, lipid, or metal ion-mediated mechanisms |
| Validation | Prioritizing based on statistical significance alone | Pursuing biologically irrelevant targets |
| Validation | Lack of orthogonal validation | False conclusions about the true mechanism of action |
Detailed Experimental Protocols for Target Deconvolution
To mitigate the pitfalls described above, robust and well-executed experimental protocols are essential. Below are detailed methodologies for key target deconvolution techniques.
Affinity-Based Pull-Down and Mass Spectrometry
This is a workhorse technique for identifying direct protein interactors from a complex biological mixture [42].
Protocol:
- Probe Design and Synthesis: Chemically modify the compound of interest to include a linker and an affinity handle (e.g., biotin). A critical control is a structurally similar but inactive analog.
- Sample Preparation: Prepare cell lysates from a relevant cell line or tissue under native conditions to preserve protein complexes.
- Affinity Enrichment: Incubate the lysate with the immobilized compound (the "bait") and the control analog. Use streptavidin-coated beads for biotinylated probes.
- Stringent Washing: Wash the beads extensively with a physiological buffer to remove non-specifically bound proteins.
- Elution and Digestion: Elute the bound proteins using a denaturing agent (e.g., SDS) or by competition with excess free compound. Digest the eluted proteins into peptides using trypsin.
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Analyze the peptides via LC-MS/MS.
- Data Analysis: Identify proteins by searching MS/MS spectra against a protein database. Compare protein abundances between the experimental bait and the control analog to identify specifically bound proteins. Statistical analysis (e.g., significance A/B tests) is used for prioritization.
Photoaffinity Labeling (PAL)
PAL is particularly valuable for capturing transient or low-affinity interactions and for studying integral membrane proteins [42].
Protocol:
- Probe Design: Synthesize a trifunctional probe containing: (a) the compound of interest, (b) a photoreactive group (e.g., diazirine), and (c) an enrichment handle (e.g., alkyne for subsequent "click chemistry" to biotin).
- Cell Treatment: Treat live cells or native lysates with the probe. Allowing the probe to bind its target in a live-cell context preserves physiological interactions.
- UV Cross-linking: Irradiate the sample with UV light (~365 nm) to activate the photoreactive group, forming a covalent bond between the probe and its target protein(s).
- Cell Lysis and "Click" Chemistry: Lyse the cells and perform a copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction to conjugate the biotin-azide tag to the alkyne-handle on the probe.
- Affinity Purification and MS: Enrich the biotinylated proteins on streptavidin beads, followed by on-bead digestion and LC-MS/MS analysis as described in section 3.1.
Cellular Thermal Shift Assay (CETSA) and Proteome-Wide Stability Profiling
This label-free method detects target engagement by measuring ligand-induced changes in protein thermal stability [43].
Protocol:
- Compound Treatment: Divide a cell lysate or intact cells into two aliquots and treat one with the compound of interest and the other with vehicle (e.g., DMSO).
- Heat Denaturation: Subject each aliquot to a series of different temperatures (e.g., from 37°C to 65°C).
- Soluble Protein Extraction: Centrifuge the heated samples to separate soluble (non-denatured) protein from insoluble (aggregated) protein.
- Proteomic Analysis: Digest the soluble fractions and analyze by LC-MS/MS. Alternatively, for a proteome-wide view (thermal proteome profiling, TPP), use isobaric tandem mass tag (TMT) labeling to multiplex all temperature points for a single LC-MS/MS run.
- Data Analysis: For each protein, plot the melting curve (soluble protein fraction vs. temperature). A significant rightward shift in the melting curve (increased melting temperature, Tm) in the compound-treated sample indicates ligand-induced stabilization and direct target engagement.
The following workflow diagram illustrates the decision-making process for selecting and applying these key methodologies.
Diagram 1: Experimental Workflow Selection.
Essential Research Reagents and Solutions
A successful target deconvolution campaign relies on a suite of specialized reagents and tools. The following table details key resources for the featured experiments.
Table 2: Key Research Reagent Solutions for Target Deconvolution
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Biotin-Azide / Alkyne Handles | Enable "click chemistry" conjugation to affinity tags (e.g., streptavidin beads) for enrichment. | Functionalizing a compound for affinity pull-down or PAL without significantly altering its core structure. |
| Photo-reactive Crosslinkers (e.g., Diazirines) | Form covalent bonds with target proteins upon UV irradiation, capturing transient interactions. | Integrating into PAL probes to "trap" the compound onto its protein target for subsequent isolation. |
| Streptavidin-Coated Magnetic Beads | Solid support for high-affinity capture and purification of biotinylated proteins and complexes. | Used in affinity pull-down and PAL workflows to isolate probe-bound proteins from a complex lysate. |
| Isobaric Tandem Mass Tags (TMT) | Multiplexing labels for LC-MS/MS that allow simultaneous quantification of proteins from multiple samples. | Enabling Thermal Proteome Profiling (TPP) by labeling soluble protein fractions from different temperatures. |
| CRISPR/Cas9 Knockout Libraries | Genome-wide screening tools to identify genes whose loss confers resistance or sensitivity to a compound. | Functional genetics approach to infer compound mechanism of action by identifying essential genetic pathways. |
| Pan-GPCR Cell Line Libraries | Collections of cell lines engineered to express individual GPCRs, enabling high-throughput screening. | Systematically testing compound activity against the "GPCRome," a therapeutically important target class [39]. |
A Path to Robust Validation: Overcoming Prioritization Pitfalls
The final and most critical phase is the rigorous validation of putative targets. This requires a multi-faceted approach that moves beyond simple identification to establish a functional link.
Table 3: Orthogonal Methods for Target Validation
| Method | Principle | What It Confirms |
|---|---|---|
| Cellular Target Engagement (e.g., CETSA) | Measure compound-induced thermal stabilization of the target protein in cells. | The compound binds to the putative target directly in a live-cell, physiological context. |
| Functional Genetics (CRISPR/i) | Knock out or knock down the putative target gene and assess impact on compound sensitivity. | The putative target is genetically required for the compound's phenotypic effect. |
| Rescue Experiments | Re-express a wild-type or compound-binding mutant of the target in knockout cells. | Re-introducing the target protein restores compound sensitivity, confirming specificity. |
| Biophysical Binding (SPR, ITC) | Measure direct binding kinetics (SPR) or thermodynamics (ITC) in a purified system. | The compound binds to the purified target with high affinity and the expected stoichiometry. |
The following diagram illustrates the integrated pathway from target identification through to robust validation, highlighting how orthogonal methods converge to provide confidence in the final target.
Diagram 2: Orthogonal Target Validation Pathway.
Target deconvolution in forward chemogenomics is a high-stakes endeavor. The pitfalls are numerous, spanning computational prediction, experimental execution, and target validation. Success is not achieved by a single experiment but through a strategic combination of orthogonal methods. A robust workflow integrates computational predictions with careful experimental design, using affinity-based methods, label-free stability profiling, and functional genomics to generate a shortlist of candidates. This must be followed by an uncompromising validation phase that confirms direct binding, cellular engagement, and functional necessity. By recognizing these common pitfalls and implementing the detailed strategies and protocols outlined in this guide, researchers can navigate the complexities of target deconvolution, de-risk their drug discovery pipelines, and unlock the full potential of phenotypic screening.
Overcoming Limitations in Reverse Chemogenomics and Selectivity Profiling
Chemogenomics represents a systematic approach to drug discovery that involves screening targeted chemical libraries against families of related drug targets, with the dual goal of identifying novel therapeutics and elucidating the functions of previously uncharacterized targets [1]. This field operates through two complementary paradigms: forward chemogenomics, which begins with a phenotypic screen to identify bioactive compounds before determining their molecular targets, and reverse chemogenomics, which starts with a specific protein target and seeks compounds that modulate its activity, subsequently characterizing the resulting phenotypes [1]. While both approaches have contributed significantly to biomedical research, reverse chemogenomics has traditionally dominated pharmaceutical discovery efforts due to its target-centric framework, which aligns with established drug development paradigms.
However, reverse chemogenomics faces substantial limitations that can hinder its effectiveness and translational success. The approach inherently depends on prior target validation, which may be incomplete or inaccurate, and often struggles with predicting cellular and organismal phenotypes from in vitro target-based data [45]. Furthermore, the selectivity profiling of compounds identified through reverse screening presents significant technical challenges, as off-target effects can lead to misleading biological interpretations and clinical failures [46]. This technical guide examines these limitations in detail and provides strategic frameworks and methodological solutions to enhance the effectiveness of reverse chemogenomics within the broader context of phenotypic drug discovery.
Critical Limitations in Reverse Chemogenomics
Fundamental Constraints of Target-First Approaches
The reverse chemogenomics paradigm, while methodologically straightforward, suffers from several inherent constraints that can limit its success in identifying therapeutically relevant compounds. First, it requires pre-existing knowledge of a target's biological function and therapeutic relevance, which for many proteins—particularly orphan targets—may be incomplete or inaccurate [45]. This approach also assumes that modulating a single target will produce a therapeutically beneficial phenotype without compensatory mechanisms or network adaptations that often occur in complex biological systems [15].
Second, there exists a fundamental disconnect between biochemical potency and cellular phenotype. A compound demonstrating excellent binding affinity and selectivity in vitro may fail to produce the desired phenotypic outcome in cellular or organismal contexts due to factors such as cellular compartmentalization, protein-protein interactions, or pathway redundancies [45]. This limitation is particularly problematic for multi-domain proteins and proteins involved in complex macromolecular assemblies, where small molecules targeting a single domain may not recapitulate the effects of genetic perturbations [15].
Selectivity Profiling Challenges
Comprehensive selectivity profiling remains a formidable challenge in reverse chemogenomics. The human proteome consists of approximately 20,000 genes, but even the most sophisticated chemogenomics libraries typically interrogate only 1,000-2,000 targets, leaving large portions of the proteome unassessed for potential off-target interactions [45]. This limited coverage creates significant blind spots in selectivity assessment.
The following table summarizes the key limitations and their experimental implications:
Table 1: Key Limitations in Reverse Chemogenomics and Their Implications
| Limitation Category | Specific Challenge | Experimental Consequence |
|---|---|---|
| Target Validation | Incomplete understanding of target function | Phenotypic outcomes may not match expectations |
| Pathway redundancy and compensatory mechanisms | Limited efficacy despite potent target engagement | |
| Selectivity Assessment | Limited proteome coverage by screening libraries | Undetected off-target effects |
| Assay conditions not reflecting cellular context | Misleading selectivity profiles | |
| Phenotypic Translation | Difficulty predicting cellular effects from biochemical data | Poor translatability between assay systems |
| Temporal aspects of target engagement | Dynamic cellular responses not captured |
Strategic Frameworks for Enhanced Reverse Chemogenomics
Integrating Forward and Reverse Approaches
A hybrid strategy that integrates elements of both forward and reverse chemogenomics can mitigate the limitations of purely target-centric approaches. This integrated framework employs phenotypic validation of targets identified through reverse chemogenomics, using chemical probes as perturbative tools to establish causal relationships between target modulation and phenotypic outcomes [47]. The systematic application of combination perturbations—mixed chemical and genetic interventions—can reveal functional relationships between pathways and help validate target engagement in biologically relevant contexts [15].
The following diagram illustrates this integrated approach:
Integrated Chemogenomics Workflow
Advanced Selectivity Profiling Methodologies
Comprehensive selectivity profiling requires orthogonal experimental approaches to overcome the limitations of any single method. A tiered profiling strategy should include:
Biophysical Methods: Techniques such as Differential Scanning Fluorimetry (DSF) can rapidly assess compound binding against panels of liability targets, including highly ligandable kinases and bromodomains whose modulation causes strong confounding phenotypes [46]. This approach was successfully implemented in the development of an NR3 nuclear receptor chemogenomics set, where DSF was used to screen candidates against ten liability targets, ensuring minimal off-target interactions [46].
Functional Cellular Assays: Panel-based profiling in cell systems expressing diverse targets provides functional context for selectivity assessment. For example, hybrid reporter gene assays across twelve nuclear receptor families demonstrated selectivity for NR3-targeted compounds, with few and non-overlapping off-target activities observed [46].
Chemical Proteomics: Methods such as affinity-based protein profiling enable untargeted exploration of compound interactions across the proteome, addressing the coverage gaps of targeted approaches [45].
Table 2: Selectivity Profiling Technologies and Their Applications
| Technology | Principle | Throughput | Key Advantage | Representative Application |
|---|---|---|---|---|
| Differential Scanning Fluorimetry (DSF) | Target thermal stability shift upon ligand binding | Medium to High | Rapid screening of predefined liability targets | NR3 CG library liability screening [46] |
| Reporter Gene Panels | Functional activity across receptor families | Medium | Physiological relevance in cellular context | NR3 CG library selectivity confirmation [46] |
| Chemical Proteomics | Affinity purification of binding proteins | Low to Medium | Proteome-wide coverage without preset targets | Identification of unknown off-targets [45] |
| Bioactivity Profiling | Multi-target screening in standardized assays | High | Quantitative comparison across target classes | Broad-scale compound annotation [48] |
Experimental Protocols for Enhanced Profiling
Comprehensive Selectivity Assessment Protocol
The following detailed protocol outlines a rigorous approach to selectivity profiling, as implemented in the development of the NR3 chemogenomics set [46]:
Step 1: Initial Compound Library Curation
- Filter annotated ligands from public databases (ChEMBL, PubChem, IUPHAR/BPS, BindingDB) based on potency (IC50/EC50 ≤ 1 μM for well-covered targets, ≤10 μM for poorly explored targets)
- Apply chemical diversity filters using pairwise Tanimoto similarity computed on Morgan fingerprints
- Prioritize compounds with diverse modes of action (agonists, antagonists, inverse agonists, modulators, degraders)
- Apply pragmatic filtering based on commercial availability and literature validation
Step 2: Tiered Selectivity Profiling
- Cytotoxicity Assessment: Evaluate compounds in HEK293T cells using multiple endpoints: growth rate, metabolic activity, and apoptosis/necrosis induction. Recommended concentration: 3-10× EC50/IC50
- Target Family Selectivity: Employ uniform hybrid reporter gene assays across diverse protein families (e.g., 12 receptors representing NR1, NR2, NR4, and NR5 families)
- Liability Panel Screening: Utilize Differential Scanning Fluorimetry against a panel of highly ligandable off-targets (e.g., 10 kinases and bromodomains with strong phenotypic effects)
Step 3: Data Integration and Compound Selection
- Apply selection criteria favoring compounds with minimal and non-overlapping off-target activities
- Optimize final set for full target family coverage with multiple chemotypes and modes of action
- Establish recommended working concentrations for each compound that ensure full on-target effect with minimal off-target activities
Phenotypic Validation Workflow
To address the target-phenotype disconnect in reverse chemogenomics, implement the following validation workflow:
Step 1: Multi-level Phenotypic Screening
- Apply selective compounds to disease-relevant in vitro models (e.g., primary cells, iPSC-derived models, 3D cultures)
- Assess phenotypes at multiple biological levels: molecular (transcriptomics, proteomics), cellular (morphology, viability, function), and pathway-specific (reporter assays, signaling readouts)
Step 2: Combination Chemical Genetics
- Systematically test compound combinations (chemical-chemical or chemical-genetic) to identify functional interactions
- Utilize orthogonal assessment methods (high-content imaging, transcriptional profiling, functional outputs)
- Apply computational tools to infer pathway relationships and network topology from interaction patterns
Step 3: Target Validation
- Employ multiple structurally distinct probes for each target to distinguish on-target from off-target effects
- Implement resistance generation and target engineering approaches to establish causal relationships
- Correlate target engagement with phenotypic responses across multiple compound concentrations and timepoints
The following diagram illustrates this comprehensive experimental workflow:
Comprehensive Experimental Workflow for Enhanced Reverse Chemogenomics
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of enhanced reverse chemogenomics requires carefully selected research reagents and tools. The following table details essential components for establishing a robust experimental framework:
Table 3: Essential Research Reagents for Advanced Chemogenomics
| Reagent Category | Specific Examples | Function & Application | Implementation Notes |
|---|---|---|---|
| Annotated Compound Libraries | NR3 CG set (34 compounds) [46], Kinase inhibitor sets | Target-focused screening with known mechanism of action | Ensure coverage of multiple chemotypes and modes of action per target |
| Liability Panels | Kinases (10), Bromodomains | Identification of confounding off-target effects | Select targets known to produce strong phenotypes when modulated |
| Cell-Based Reporter Systems | Uniform hybrid reporter assays for nuclear receptors [46] | Functional selectivity assessment in cellular context | Standardize across target families for comparable data |
| Phenotypic Screening Platforms | High-content imaging, 3D culture systems, iPSC-derived models | Translation of target modulation to phenotypic outcomes | Use disease-relevant models with physiological expression patterns |
| Proteomic Profiling Tools | Affinity matrices, activity-based probes | Untargeted exploration of compound interactions | Complementary to targeted approaches for comprehensive coverage |
Reverse chemogenomics remains a powerful approach for targeted drug discovery, but its limitations necessitate strategic enhancements in selectivity profiling and phenotypic validation. By implementing integrated forward-reverse approaches, comprehensive multi-tiered selectivity assessment, and rigorous phenotypic validation, researchers can significantly improve the success rate of target-based discovery efforts. The experimental frameworks and protocols outlined in this technical guide provide a roadmap for overcoming the key limitations of traditional reverse chemogenomics, ultimately facilitating the identification of more efficacious and specific therapeutic agents.
Future directions in the field will likely involve increased integration of artificial intelligence and machine learning approaches to predict selectivity profiles and compound-target interactions [49] [4]. Additionally, the development of more sophisticated disease models and high-content phenotypic readouts will further bridge the gap between target engagement and functional therapeutic effects. As these technologies mature, the distinction between forward and reverse chemogenomics may increasingly blur, yielding hybrid approaches that leverage the strengths of both paradigms to accelerate therapeutic discovery.
The Role of AI and Machine Learning in Predicting Drug-Target Interactions
The process of drug discovery has traditionally been characterized by high costs, lengthy timelines, and high failure rates, often taking over a decade and costing billions of dollars to bring a new therapeutic to market. [50] [51] At the heart of this process lies the critical challenge of understanding drug-target interactions (DTIs)—the complex interplay between pharmaceutical compounds and their biological targets. Artificial intelligence (AI) and machine learning (ML) have emerged as transformative technologies in this domain, enabling researchers to rapidly identify and optimize potential drug candidates with unprecedented efficiency. [52] [53]
This technological revolution is fundamentally reshaping chemogenomics—the systematic study of targeted chemical libraries against families of drug targets. [1] AI-powered DTI prediction sits at the intersection of two complementary chemogenomics approaches: forward chemogenomics, which seeks molecules that produce a desired phenotype before identifying the molecular target, and reverse chemogenomics, which starts with a specific protein target and searches for compounds that modulate its activity. [1] [11] By leveraging massive chemical and biological datasets, ML models can now accelerate both paradigms, compressing discovery timelines that previously required years of experimental work into months or even weeks. [21]
This technical guide examines the core methodologies, experimental frameworks, and practical implementations of AI in DTI prediction, with a specific focus on its role in advancing modern chemogenomics research for drug development professionals and computational biologists.
Chemogenomics Framework and AI Integration
Forward vs. Reverse Chemogenomics
Chemogenomics provides a systematic framework for drug discovery by exploring the interaction space between chemical compounds and biological targets. [1] The two principal approaches create distinct discovery pathways:
Forward Chemogenomics: This phenotype-first approach begins with screening compounds against cellular or organismal models to identify molecules that induce a desired phenotypic change. [1] The molecular targets responsible for the phenotype are identified subsequently, making this method particularly valuable for investigating complex biological systems where mechanisms of action are unknown. AI algorithms enhance this approach by analyzing high-content screening data and predicting potential target-phenotype relationships from complex multivariate readouts. [21]
Reverse Chemogenomics: This target-first approach begins with a specific, well-characterized protein target and screens for compounds that selectively modulate its activity. [1] This strategy benefits from clearly defined structure-activity relationships but may overlook complex polypharmacological effects. ML models excel in this domain by leveraging known target-ligand interactions to predict novel binders through similarity-based reasoning and structural analysis. [11]
AI-Driven Unification of Chemogenomic Approaches
Modern AI platforms are increasingly blurring the distinction between forward and reverse chemogenomics by creating integrated systems that leverage the strengths of both approaches. [21] For instance, companies like Recursion and Exscientia have merged phenotypic screening with target-focused design, creating unified discovery engines that can navigate from phenotypic observations to optimized chemical matter seamlessly. [21] The merger of Recursion's extensive phenomic screening capabilities with Exscientia's automated precision chemistry represents a prime example of this convergence, creating an "AI drug discovery superpower" that operates across both chemogenomic paradigms. [21]
Table 1: AI Platforms Exemplifying Integrated Chemogenomic Approaches
| Company/Platform | Primary Approach | Key Technology | Clinical Stage Examples |
|---|---|---|---|
| Exscientia | Reverse Chemogenomics | Generative Chemistry + Automated Design-Make-Test Cycles | CDK7 inhibitor (GTAEXS-617) in Phase I/II trials [21] |
| Recursion | Forward Chemogenomics | Phenomic Screening + Computer Vision | Multiple programs in oncology and neurology [21] |
| Insilico Medicine | Hybrid Approach | Generative Adversarial Networks (GANs) | ISM001-055 for IPF (Phase IIa) [21] |
| Schrödinger | Reverse Chemogenomics | Physics-Based Simulation + ML | TAK-279 (TYK2 inhibitor) in Phase III [21] |
| BenevolentAI | Knowledge-Driven | Knowledge Graph Mining | Multiple candidates in clinical stages [21] |
AI Methodologies for Drug-Target Interaction Prediction
Computational Foundations
AI-based DTI prediction encompasses diverse computational strategies that can be categorized by their underlying methodology and data requirements:
Structure-Based Approaches: These methods, including molecular docking and molecular dynamics simulations, rely on the 3D structural information of target proteins to predict binding interactions and affinities. [52] [11] While powerful, these approaches require high-quality structural data and significant computational resources, limiting their application to targets with known or reliably modeled structures.
Ligand-Based Approaches: These methods, including quantitative structure-activity relationship (QSAR) modeling, predict DTIs by comparing candidate compounds to known active molecules for a specific target. [52] [11] Their predictive power depends heavily on the availability of known ligands for the target of interest.
Network-Based Approaches: These methods construct heterogeneous networks integrating diverse data types (chemical, genomic, proteomic, pharmacological) and use graph algorithms to infer novel interactions based on network topology and similarity measures. [52] [54]
Machine Learning-Based Approaches: These methods extract latent features from chemical and biological data to build predictive models for binary interaction classification or binding affinity regression. [52] [54] They have gained prominence due to their ability to integrate multimodal data and generalize across diverse target families.
Deep Learning Architectures
Deep learning has emerged as a particularly powerful paradigm for DTI prediction, with several specialized architectures demonstrating state-of-the-art performance:
Convolutional Neural Networks (CNNs): Applied to molecular representations such as SMILES strings or molecular graphs to learn hierarchical features predictive of binding activity. [52] [53] For example, DeepDTA uses CNN architectures to learn representations from SMILES strings of compounds and amino acid sequences of proteins. [52]
Graph Neural Networks (GNNs): Operate directly on molecular graphs, capturing both structural topology and atomic properties to generate enriched molecular representations. [55] [54] Message-passing neural networks (MPNNs) have shown particular success in predicting molecular properties relevant to drug-target binding. [55]
Transformer Models: Leverage self-attention mechanisms to capture long-range dependencies in protein sequences and molecular structures. [52] [54] Recent transformer-based models like TransformerCPI have demonstrated superior performance in DTI prediction tasks. [52]
Multi-Modal Learning Architectures: Integrate diverse data types (sequences, structures, interaction networks) through specialized encoding pathways that are fused for joint prediction. The DTIAM framework exemplifies this approach, using separate pre-training modules for drugs and targets before integrating them for interaction prediction. [52]
Advanced Frameworks: DTIAM Case Study
The DTIAM framework represents a cutting-edge approach that addresses multiple limitations of previous DTI prediction methods through unified self-supervised learning. [52] Its architecture consists of three specialized modules:
Drug Molecular Pre-training Module: Takes molecular graphs as input, segments them into substructures, and learns representations through multiple self-supervised tasks including masked language modeling, molecular descriptor prediction, and functional group prediction. [52]
Target Protein Pre-training Module: Uses transformer attention maps to learn representations and contacts from large amounts of protein sequence data through unsupervised language modeling. [52]
Drug-Target Prediction Module: Integrates compound and protein representations using machine learning models within an automated framework that utilizes multi-layer stacking and bagging techniques. [52]
This architecture demonstrates substantial performance improvements over previous methods, particularly in challenging cold-start scenarios where either the drug or target lacks known interactions in training data. [52]
Diagram: DTIAM Unified Framework for DTI Prediction
Experimental Protocols and Methodologies
Standardized Workflows for DTI Prediction
Implementing robust AI-driven DTI prediction requires carefully designed experimental protocols that address data collection, model training, validation, and interpretation. The following workflow outlines a comprehensive methodology suitable for both forward and reverse chemogenomics applications:
Phase 1: Data Collection and Curation
- Compound Libraries: Curate chemical structures from databases such as ChEMBL, PubChem, or ZINC15, ensuring appropriate representation of relevant chemical space. [52] [54] Standardize structures, remove duplicates, and check for validity using toolkits like RDKit. [55]
- Target Information: Collect protein sequences from UniProt, structural data from PDB (where available), and functional annotations from Gene Ontology or similar resources. [54]
- Interaction Data: Gather known DTIs from public databases like BindingDB, Davis, or KIBA, paying careful attention to experimental conditions and measurement types. [52] [54]
- Data Partitioning: Implement appropriate train/validation/test splits that reflect real-world scenarios, including warm start (random split), drug cold start (new drugs), and target cold start (new targets). [52]
Phase 2: Feature Representation
- Molecular Featurization: Convert chemical structures into numerical representations using extended-connectivity fingerprints (ECFPs), molecular descriptors, graph representations, or learned embeddings from self-supervised pre-training. [52] [51]
- Protein Featurization: Represent protein targets through sequence embeddings (e.g., from protein language models), structural features (when structures are available), or evolutionary information from multiple sequence alignments. [52]
- Feature Integration: Develop joint representation spaces that capture complementary information from both compounds and targets, using early fusion (feature concatenation), late fusion (separate encoders with combined prediction), or cross-attention mechanisms. [52]
Phase 3: Model Training and Validation
- Algorithm Selection: Choose appropriate ML architectures based on data characteristics and prediction tasks—binary classification for interaction prediction, regression for affinity estimation, or multi-task learning for parallel prediction of multiple properties. [52] [53]
- Training Protocol: Implement appropriate regularization strategies (dropout, weight decay), optimization methods (Adam, SGD with momentum), and learning rate schedules to prevent overfitting. [53]
- Validation Framework: Employ rigorous cross-validation strategies that match the intended use case, with particular attention to temporal validation (time-split test sets) and cold-start scenarios for maximal real-world relevance. [52] [56]
- Performance Metrics: Select evaluation metrics aligned with application goals—AUROC and AUPRC for binary classification, RMSE and Pearson correlation for affinity prediction, with special attention to performance in cold-start scenarios. [52] [53]
Prospective Validation and Experimental Confirmation
The ultimate test of any DTI prediction model lies in its prospective performance on truly novel interactions. The following protocol outlines a robust framework for experimental confirmation:
Computational Screening: Apply trained models to screen virtual compound libraries against targets of interest, prioritizing candidates based on predicted activity and confidence metrics. [52]
Diversity Selection: Select compounds for testing that represent both chemically diverse scaffolds and varying prediction confidence levels to properly assess model performance across chemical space. [56]
Experimental Testing: Validate top predictions using appropriate biochemical or cellular assays, ensuring assay conditions match the training data context where possible. [52]
Iterative Refinement: Use experimental results to retrain and improve models, potentially incorporating active learning strategies to maximize information gain from limited experimental resources. [56]
A notable example of successful prospective validation comes from the DTIAM framework, which identified effective TMEM16A inhibitors from a high-throughput molecular library of 10 million compounds, with subsequent confirmation via whole-cell patch clamp experiments. [52]
Table 2: Key Research Reagents and Computational Tools for AI-Driven DTI Prediction
| Category | Tool/Resource | Primary Function | Application Context |
|---|---|---|---|
| Chemical Databases | PubChem, ChEMBL, ZINC | Source of chemical structures and bioactivity data | Compound library construction for virtual screening [55] [54] |
| Protein Databases | UniProt, PDB, AlphaFold DB | Protein sequences, structures, and annotations | Target featurization and structural modeling [52] [54] |
| Interaction Databases | BindingDB, Davis, KIBA | Known drug-target interactions and affinity measurements | Model training and benchmarking [52] [54] |
| Cheminformatics Tools | RDKit, DeepChem | Molecular manipulation, featurization, and property calculation | Chemical data preprocessing and feature generation [55] |
| Deep Learning Frameworks | PyTorch, TensorFlow | Neural network implementation and training | Custom model development and experimentation [52] |
| Specialized Platforms | DTIAM, DeepDTA, MONN | End-to-end DTI prediction | Benchmarking and production prediction pipelines [52] |
Signaling Pathways and Mechanistic Predictions
Beyond predicting mere binding events, advanced AI systems are increasingly capable of elucidating mechanisms of action (MoA), including distinguishing between activation and inhibition—a critical distinction in therapeutic development. [52] This capability represents a significant advancement in predictive modeling, moving beyond simple binding prediction to functional outcome assessment.
Diagram: MoA Prediction from DTI
The DTIAM framework specifically addresses this challenge by incorporating MoA prediction as a core capability, distinguishing activation from inhibition mechanisms through multi-task self-supervised pre-training that captures subtle structural and contextual determinants of functional outcomes. [52] This capability is particularly valuable in forward chemogenomics applications, where phenotypic screening identifies compounds with desired effects, but the specific molecular mechanisms remain unknown.
Implementation Challenges and Future Directions
Despite significant progress, AI-driven DTI prediction faces several persistent challenges that represent active research frontiers:
Data Quality and Standardization
The performance of ML models heavily depends on training data quality, yet biomedical data often suffers from inconsistency, experimental noise, and systematic biases. [56] A critical analysis comparing IC50 values for the same compounds across different laboratories found "almost no correlation between the reported values from different papers," highlighting the profound standardization challenges in the field. [56] Initiatives like OpenADMET aim to address this through centralized generation of high-quality, standardized datasets specifically designed for ML model development. [56]
Generalization and Transfer Learning
The "cold start" problem—predicting interactions for novel drugs or targets with no known interactions—remains a significant challenge. [52] Transfer learning and self-supervised pre-training approaches, such as those used in DTIAM, show promise in addressing this limitation by learning generalizable representations from large unlabeled datasets before fine-tuning on specific prediction tasks. [52] Foundation models pre-trained on massive chemical and biological corpora are emerging as powerful tools for improving generalization across diverse target families and chemical spaces. [56]
Interpretability and Mechanistic Insight
While deep learning models often achieve high predictive accuracy, their "black box" nature can limit mechanistic insights crucial for drug optimization. [50] Incorporating explainable AI techniques, attention mechanisms, and leveraging structural biology insights can help address this limitation by identifying key molecular determinants of binding and function. [52] [56] For instance, MONN uses non-covalent interactions as additional supervision to guide the model to capture key binding sites, enhancing interpretability. [52]
Emerging Paradigms and Future Outlook
The field is rapidly evolving toward more integrated and sophisticated approaches:
- Generative AI: The application of generative adversarial networks (GANs) and variational autoencoders (VAEs) enables de novo design of novel compounds with optimized target interaction profiles, moving beyond virtual screening to generative chemistry. [21] [53]
- Large Language Models: Protein and molecular language models trained on massive sequence and structural datasets are demonstrating remarkable capabilities in capturing complex biological principles relevant to DTI prediction. [54]
- Multimodal Integration: Combining diverse data types—chemical, genomic, proteomic, structural, and clinical—within unified modeling frameworks promises more comprehensive and predictive representations of drug-target relationships. [54]
- Quantum Chemistry Integration: Incorporating quantum mechanical calculations into ML frameworks offers potential for more accurate modeling of molecular interactions and reaction mechanisms relevant to drug-target binding. [54]
AI and machine learning have fundamentally transformed the prediction of drug-target interactions, enabling both forward and reverse chemogenomics approaches to operate at unprecedented scale and efficiency. Frameworks like DTIAM demonstrate how self-supervised learning and multimodal integration can address longstanding challenges in generalization and mechanistic prediction. [52] The convergence of high-quality data generation initiatives, advanced algorithmic approaches, and closer integration with experimental validation creates a virtuous cycle of improvement in predictive accuracy and biological relevance.
As these technologies continue to mature, fully ML-integrated drug discovery pipelines will increasingly define the future of pharmaceutical development, compressing timelines, reducing costs, and ultimately delivering better therapeutics to patients. For researchers and drug development professionals, mastery of these AI methodologies is no longer optional but essential for remaining at the forefront of modern drug discovery science.
Leveraging Open-Source Data and Initiatives like EUbOPEN and Target 2035
The escalating costs and high failure rates associated with conventional drug discovery have necessitated a paradigm shift toward more efficient, data-driven approaches. Chemogenomics represents one such strategic evolution, systematically investigating the interaction between small molecules and biological target families on a genome-wide scale [1]. This field operates primarily through two complementary paradigms: forward chemogenomics, which begins with a phenotypic observation and seeks to identify the responsible molecular target, and reverse chemogenomics, which starts with a specific protein target and searches for compounds that modulate its activity [1] [12]. The ultimate goal of chemogenomics is the parallel identification of novel drug targets and their biologically active modulators [1].
Global initiatives like Target 2035 and EUbOPEN are fundamentally underpinned by these chemogenomic principles. Target 2035 is an international open science initiative with the ambitious goal of developing chemical or biological modulators for nearly all human proteins by the year 2035 [8]. The EUbOPEN (Enabling and Unlocking Biology in the OPEN) consortium, a public-private partnership funded by the Innovative Medicines Initiative, is a major contributor to this goal. Its mission is to create, distribute, and annotate the largest openly available set of high-quality chemical modulators for human proteins, including a chemogenomic library covering approximately one-third of the druggable proteome and at least 100 high-quality, open-access chemical probes [57] [8]. This whitepaper provides a technical guide for leveraging these resources within forward and reverse chemogenomics research frameworks.
Core Concepts: Forward vs. Reverse Chemogenomics
The distinction between forward and reverse chemogenomics lies in the starting point of the investigation and has profound implications for the experimental workflow, required tools, and data interpretation strategies.
Forward (Classical) Chemogenomics
In forward chemogenomics, research begins with the observation of a desired phenotype in a cell-based or organism-based assay. The molecular basis for this phenotype is initially unknown [1]. The subsequent challenge is to deconvolute the protein target(s) responsible for the observed phenotypic effect.
- Objective: To identify the molecular target and mechanism of action (MOA) of a small molecule that produces a specific phenotypic response [12].
- Typical Workflow: A phenotypic screen (e.g., inhibition of tumor growth) identifies active compounds. These modulators are then used as tools to identify the responsible protein [1].
- Key Challenge: Designing phenotypic assays that facilitate a direct path from screening to target identification [1].
Reverse Chemogenomics
Reverse chemogenomics adopts a target-centric approach. It begins with a specific, well-defined protein target believed to be therapeutically relevant and aims to find compounds that perturb its function.
- Objective: To discover small molecules that interact with a predefined protein target and to characterize the resulting phenotype [1].
- Typical Workflow: Small molecules are screened for activity against a purified target in an in vitro assay. Hits are then tested in cellular or whole-organism models to analyze the induced biological response [1] [12].
- Advantage: The target is known from the outset, simplifying the initial stages of discovery.
The following diagram illustrates the logical flow and key differences between these two fundamental approaches.
The Open-Source Landscape: Key Initiatives and Data Repositories
Leveraging chemogenomics requires access to high-quality, annotated chemical and biological data. Several pivotal initiatives and public repositories provide the foundational resources for this research.
The EUbOPEN Consortium
EUbOPEN is a 5-year project with a total budget of €65.8 million, aiming to systematically generate and characterize open-access chemical tools [57] [8]. Its outputs are structured around four pillars, detailed in the table below.
Table 1: Strategic Pillars and Outputs of the EUbOPEN Consortium
| Pillar of Activity | Key Objectives | Outputs & Deliverables |
|---|---|---|
| Chemogenomic Library Collection | Assemble an open-access chemogenomic library (~5,000 compounds) covering ~1,000 proteins (1/3 of druggable proteome) [57] [8]. | Well-annotated compound sets for target families like kinases, GPCRs, E3 ligases, and SLCs, profiled in selectivity panels [8]. |
| Chemical Probe Discovery | Synthesize ≥100 high-quality, open-access chemical probes and negative controls [57] [8]. | Peer-reviewed probes meeting strict criteria (e.g., potency <100 nM, >30-fold selectivity) [8]. |
| Disease-Relevant Profiling | Disseminate reliable protocols for ≥20 primary patient cell-based assays [57]. | Data from profiling compounds in disease-relevant assays (e.g., inflammatory bowel disease, cancer) [57] [8]. |
| Data & Reagent Dissemination | Establish infrastructure and governance for data/reagent sharing [57]. | Public data repositories; distribution of >6,000 probe samples without restrictions [8]. |
Target 2035 is the overarching global initiative that EUbOPEN supports. Its goal is to create pharmacological modulators for most human proteins by 2035, providing peer-reviewed tools and data freely to the research community [8].
For researchers, several public databases are indispensable for chemogenomics studies:
- PubChem: A foundational NIH resource containing 119 million compounds, 295 million bioactivities, and integrated data from over 1,000 sources, including drug information, toxicity data, and natural product annotations [58].
- Pharos: Provides target development levels and is integrated into PubChem, helping prioritize less-studied "dark" targets [58] [27].
- ChEMBL, DrugBank, ZINC15: Other critical databases for chemical structures, drug data, and commercially available compounds for virtual screening [59].
Experimental Methodologies and Protocols
This section details standard experimental protocols for conducting chemogenomics research, from initial screening to target identification and validation.
Phenotypic Screening & Hit Identification (Forward Chemogenomics)
Objective: To identify small molecules that induce a specific phenotypic change in a cellular or organismal model.
Protocol:
- Assay Development: Establish a robust, disease-relevant phenotypic assay (e.g., high-content imaging of cell morphology, reporter gene assay, or growth inhibition in a patient-derived cell line) [12].
- Compound Library Selection: Screen a curated library. The EUbOPEN chemogenomic library is ideal for this purpose, as compounds have known but overlapping target profiles, facilitating later deconvolution [8].
- Screening Execution: Conduct the screen in a high-throughput or high-content format. Include appropriate controls (e.g., DMSO vehicle and a known positive control).
- Hit Validation: Confirm primary hits through dose-response experiments to determine potency (IC50/EC50) and assess preliminary cytotoxicity to ensure a reasonable therapeutic window [8].
Target Deconvolution Methods (Forward Chemogenomics)
Once a phenotypic hit is confirmed, the critical step of target identification begins. The following table compares the primary methods.
Table 2: Key Target Deconvolution Methods in Forward Chemogenomics
| Method | Principle | Workflow Summary | Advantages | Limitations/Downsides |
|---|---|---|---|---|
| Affinity Purification | Immobilize the bioactive compound and use it as bait to pull down direct binding partners from a cell lysate [12]. | 1. Synthesize a functionalized analog (e.g., with biotin). 2. Incubate with cell lysate. 3. Capture binding proteins on streptavidin beads. 4. Wash and elute proteins. 5. Identify proteins by mass spectrometry [12]. | Most direct method; can identify protein complexes; uses human proteins [12]. | Requires compound immobilization without losing activity; high background from nonspecific binding; control beads are critical [12]. |
| Genetic Interaction Profiling | Measure the sensitivity of a collection of gene-deletion or gene-knockdown strains to the compound [19]. | 1. Use a barcoded yeast deletion collection (YKO) or a mammalian gene knockdown library (e.g., CRISPRi). 2. Grow the pooled library with/without the compound. 3. Quantify strain abundance via barcode sequencing. 4. Sensitive or resistant strains indicate genetic interaction and suggest target pathway [19]. | Genome-wide and unbiased; can reveal entire pathway; does not require protein purification [19]. | May not directly identify the binding target; limited to model organisms for some libraries; can identify downstream effects [19]. |
| Haploinsufficiency Profiling (HIP) | In a heterozygous deletion strain, a 50% reduction in the target protein level can confer hypersensitivity to a compound targeting that protein [19]. | 1. Use a pooled heterozygous yeast deletion library. 2. Perform competitive growth assay with the compound. 3. The strain with the deleted allele of the drug target will be underrepresented in the pool [19]. | Can directly identify the protein target in a single experiment [19]. | Primarily applicable to haploid organisms like yeast; not all targets show a haploinsufficient phenotype [19]. |
| Computational Inference | Compare the compound's biological profile (e.g., transcriptomic, cytological) to reference profiles in large databases [19] [12]. | 1. Generate a signature for the query compound (e.g., gene expression profile from RNA-seq). 2. Query a reference database (e.g., LINCS L1000). 3. Identify reference compounds with the most similar signatures. 4. Infer that the query compound shares the MOA/target of the best-matching reference compounds [19]. | Low cost; uses existing data; can provide immediate mechanistic hypotheses [28]. | Relies on completeness of reference database; inferences are indirect and require experimental validation; "guilt-by-association" can be misleading [19] [28]. |
The following workflow diagram integrates these methods into a coherent target deconvolution strategy.
In Vitro Screening & Hit-to-Lead Optimization (Reverse Chemogenomics)
Objective: To identify and optimize compounds that bind to a predefined, purified protein target.
Protocol:
- Target Production & Assay Development: Express and purify the recombinant target protein. Develop a robust in vitro biochemical assay (e.g., fluorescence-based, radiometric) to measure its activity.
- Virtual Screening: Before experimental screening, use computational methods to prioritize compounds from large virtual libraries (e.g., ZINC15, EUbOPEN virtual libraries). This involves:
- Ligand-Based Virtual Screening (LBVS): If known active compounds exist, use molecular fingerprints and machine learning to find structurally similar compounds [59].
- Structure-Based Virtual Screening (SBVS): If a 3D protein structure is available, use molecular docking to computationally predict binding poses and affinities of compounds from a library [59].
- High-Throughput Screening (HTS): Experimentally screen a physical compound library (e.g., the EUbOPEN CG library) using the biochemical assay.
- Hit Validation & Selectivity Screening: Confirm HTS and virtual screening hits. Test confirmed hits against related target family members (e.g., a kinase panel) to assess initial selectivity, a key step in developing a quality chemical probe [8].
- Lead Optimization: Use iterative cycles of medicinal chemistry to optimize hit compounds based on potency, selectivity, and drug-like properties (e.g., solubility, permeability). Cheminformatics tools like RDKit are essential for calculating molecular descriptors and analyzing structure-activity relationships (SAR) [59] [60].
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful execution of chemogenomics protocols requires a suite of reliable software tools and chemical resources.
Table 3: Essential Cheminformatics and Chemical Tools for Chemogenomics
| Tool/Resource Name | Type | Primary Function in Chemogenomics |
|---|---|---|
| RDKit | Open-source Cheminformatics Library [60] | Molecule drawing, descriptor calculation, chemical fingerprint generation, and SAR analysis via a Python API [59] [60]. |
| Chemistry Development Kit (CDK) | Open-source Cheminformatics Library [60] | Similar to RDKit, provides chemical structure representation, descriptor calculation, and supports various file formats [60]. |
| Open Babel | Chemical Toolbox [60] | Crucial for format conversion, structure searching, and manipulation of chemical structures from different databases. |
| PaDEL-Descriptor | Descriptor Calculation Software [60] | Calculates a comprehensive set of molecular descriptors for QSAR modeling and property prediction. |
| EUbOPEN Chemogenomic Library | Physical Compound Collection [57] [8] | A pre-annotated set of ~5,000 compounds for screening; ideal for phenotypic screens and building initial structure-activity relationships. |
| EUbOPEN Chemical Probes | Physical Compound Collection [8] | High-quality, selective tool compounds for target validation and as positive controls in assays. |
| PubChem | Public Database [58] | Primary resource for accessing bioactivity data, chemical structures, and links to other targets and pathways. |
The integration of open-source data and initiatives like EUbOPEN and Target 2035 provides an unprecedented foundation for advancing drug discovery through chemogenomics. By understanding and applying the distinct yet complementary workflows of forward and reverse chemogenomics, researchers can systematically illuminate the links between chemical compounds, their protein targets, and phenotypic outcomes. The availability of high-quality, openly accessible chemical probes, chemogenomic libraries, and powerful public databases empowers the global scientific community to accelerate the exploration of the druggable genome and ultimately translate these findings into new therapeutic strategies for human disease.
Best Practices for Data Integration from Cheminformatics and Bioinformatics
The convergence of cheminformatics and bioinformatics has become a critical enabler in modern drug discovery, particularly within the framework of chemogenomics. Chemogenomics represents a systematic approach to interrogating the interactions between chemical compounds and biological target families, with the ultimate goal of identifying novel drugs and drug targets [1]. This discipline operates on the principle that a comprehensive understanding of the ligand-target interaction space can accelerate the discovery process for entire protein families [6].
The strategic importance of data integration is fundamentally shaped by two complementary research paradigms: forward chemogenomics and reverse chemogenomics. In forward chemogenomics (also termed classical chemogenomics), researchers identify compounds that induce a specific phenotypic response in cells or whole organisms and subsequently work to identify the specific protein targets responsible for this phenotype [1] [6]. Conversely, reverse chemogenomics begins with a specific protein target and screens for small molecules that modulate its activity, then analyzes the phenotypic consequences of this interaction to validate biological function [1] [35]. Both approaches require sophisticated integration of chemical and biological data, albeit with different starting points and analytical workflows.
Effective data integration bridges the chemical space (comprising compound structures, properties, and activities) with the biological space (encompassing genomic sequences, protein structures, and phenotypic responses). This synergy enables researchers to build predictive models that can anticipate novel drug-target interactions, identify potential off-target effects, and facilitate drug repurposing efforts [6] [11]. As the volume and complexity of chemical and biological data continue to grow exponentially, establishing robust, standardized practices for data integration has become indispensable for advancing chemogenomics research.
Foundational Concepts and Data Domains
Successful integration requires a clear understanding of the distinct yet complementary data domains involved. Cheminformatics focuses primarily on the chemical space, dealing with small molecules and their properties, while bioinformatics addresses the biological space, focusing on macromolecules, pathways, and systems.
Cheminformatics Data Domain
The cheminformatics domain centers on the systematic management and analysis of chemical compound information. Key components include:
- Chemical Structures: Represented using standardized notations such as SMILES (Simplified Molecular Input Line Entry System), InChI (International Chemical Identifier), or molecular graph representations [59] [61]. These notations enable computational manipulation and similarity assessment between compounds.
- Molecular Descriptors: Quantitative representations of molecular properties including physicochemical characteristics (e.g., molecular weight, logP, polar surface area), electronic properties, and topological indices [59]. These descriptors serve as critical inputs for quantitative structure-activity relationship (QSAR) models and machine learning algorithms.
- Chemical Libraries: Structured collections of compounds, either physically available or virtual, annotated with associated data. These include targeted libraries focused on specific protein families (e.g., kinases, GPCRs) and diverse libraries designed to explore broad chemical space [1] [6].
- Compound-Target Interactions: Data on binding affinities (IC50, Ki), functional activities (EC50), and selectivity profiles against various biological targets [6].
The preprocessing and standardization of chemical data are essential preliminary steps. This involves removing duplicates, correcting errors, standardizing formats, and generating consistent molecular representations to ensure data quality and interoperability [59].
Bioinformatics Data Domain
The bioinformatics domain encompasses the biological context in which compounds exert their effects. Essential data types include:
- Genomic Data: DNA and RNA sequence information, including single nucleotide polymorphisms (SNVs), insertions/deletions (indels), and structural variants [62]. The reference genome build hg38 is now recommended as the standard for clinical bioinformatics applications [62].
- Protein Information: Amino acid sequences, three-dimensional structures (from X-ray crystallography, cryo-EM, or computational prediction tools like AlphaFold2), and functional annotations [63].
- Pathway and Network Data: Information about biological pathways, protein-protein interaction networks, and gene regulatory networks that provide context for understanding compound mechanisms of action [11].
- Phenotypic Data: High-content screening data, gene expression profiles, and clinical readouts that capture the biological effects of compound treatment [1].
The expansion of public databases such as PubChem, ChEMBL, and various genomic data repositories has dramatically increased the accessibility of both chemical and biological data, facilitating more comprehensive integration efforts [61].
Table 1: Core Data Types in Cheminformatics and Bioinformatics Integration
| Data Domain | Data Type | Description | Common Formats/Sources |
|---|---|---|---|
| Cheminformatics | Chemical Structures | 2D/3D molecular representations | SMILES, InChI, MOL files [59] [61] |
| Molecular Descriptors | Quantitative properties of compounds | Physicochemical properties, molecular fingerprints [59] | |
| Compound Libraries | Collections of annotated compounds | PubChem, ZINC15, DrugBank [59] [6] | |
| Bioactivity Data | Measurements of compound-target interactions | IC50, Ki, EC50 values [6] | |
| Bioinformatics | Genomic Sequences | DNA/RNA sequence information | FASTA, FASTQ, BAM [62] |
| Protein Structures | 3D structural information | PDB files, AlphaFold models [63] | |
| Variant Data | Genetic variations | VCF files with SNVs, indels, CNVs [62] | |
| Pathway Information | Biological pathways and networks | BioPAX, SBML, KEGG [11] |
Data Integration Methodologies and Workflows
The integration of cheminformatics and bioinformatics data follows distinct methodological pathways aligned with forward and reverse chemogenomics approaches. Below, we detail protocols and workflows for each paradigm.
Data Integration for Forward Chemogenomics
Forward chemogenomics begins with phenotypic screening and progresses toward target identification. The data integration workflow supports this progression by connecting phenotypic observations to molecular targets.
Experimental Protocol: Phenotype-Driven Target Deconvolution
Phenotypic Screening Implementation
- Objective: Identify compounds inducing a phenotype of interest (e.g., inhibition of cancer cell growth, alteration of microbial viability).
- Methods: Conduct high-throughput phenotypic screening using cell-based or whole-organism assays. The NCI60 cancer cell line panel represents a well-established example where compound cytotoxicity patterns across multiple cell lines generate distinctive phenotypic fingerprints [35].
- Data Collection: Quantify phenotypic responses using high-content imaging, viability assays, or other relevant readouts. Record dose-response relationships for hit compounds.
Chemoinformatic Analysis of Active Compounds
- Objective: Identify structural and physicochemical patterns among active compounds.
- Methods:
- Calculate molecular descriptors and fingerprints for all screened compounds [59].
- Perform chemical similarity analysis using tools like RDKit or Open Babel to cluster active compounds and identify common chemotypes [59].
- Apply machine learning methods to build models that distinguish active from inactive compounds based on chemical features.
Bioinformatic Target Hypothesis Generation
- Objective: Generate plausible target hypotheses for follow-up validation.
- Methods:
- Query chemogenomic databases (e.g., ChEMBL, BindingDB) to identify known targets of structurally similar compounds [6] [11].
- Use similarity-based inference methods such as the "similarity principle" – the premise that structurally similar compounds are likely to share molecular targets [1] [35].
- Implement target prediction algorithms that leverage chemical similarity to known ligands alongside target sequence similarity [6].
Experimental Target Validation
- Objective: Confirm the interaction between candidate compounds and proposed targets.
- Methods:
- Conduct direct binding assays (e.g., surface plasmon resonance, thermal shift assays) for top target hypotheses.
- Use genetic approaches (e.g., CRISPR knockouts, RNA interference) to assess whether target modulation reproduces the phenotypic effect.
- Perform functional studies to establish mechanistic relationships between target engagement and phenotype.
The following diagram illustrates the integrated data flow in forward chemogenomics:
Data Integration for Reverse Chemogenomics
Reverse chemogenomics adopts a target-centric approach, beginning with a specific protein of interest and progressing toward phenotype understanding. The data integration workflow supports target-based screening and phenotypic contextualization.
Experimental Protocol: Target-Driven Phenotype Elucidation
Target Selection and Characterization
- Objective: Select and characterize a therapeutic target protein.
- Methods:
- Perform sequence analysis and family classification to place the target within its phylogenetic context [6].
- Determine or model the three-dimensional protein structure using experimental methods (X-ray crystallography, cryo-EM) or computational approaches (AlphaFold2, homology modeling) [63].
- Identify binding sites, functional domains, and known ligand interactions through structural bioinformatics analysis.
Structure-Based Virtual Screening
- Objective: Identify potential ligand molecules that interact with the target.
- Methods:
- Prepare a virtual compound library for screening, applying drug-likeness filters (e.g., Lipinski's Rule of Five) and optimizing molecular representations [59].
- Perform molecular docking using tools such as GROMACS or AutoDock to predict binding poses and affinities of compounds against the target structure [63] [59].
- Use consensus scoring approaches to rank compounds by their predicted binding energy and interaction quality.
Experimental Screening and Validation
- Objective: Experimentally test predicted compounds and validate target engagement.
- Methods:
- Conduct in vitro binding or functional assays for top-ranked virtual hits.
- Determine potency (IC50/EC50) and efficacy of confirmed hits.
- Perform selectivity profiling against related targets to assess specificity.
Phenotypic Contextualization
- Objective: Understand the cellular and organismal consequences of target modulation.
- Methods:
- Test active compounds in cell-based assays relevant to the target's proposed biological function.
- Use omics technologies (transcriptomics, proteomics) to characterize compound-induced changes at a systems level.
- Correlate target engagement with phenotypic outcomes to establish therapeutic relevance.
The following diagram illustrates the integrated data flow in reverse chemogenomics:
Unified Data Integration Platforms
Both forward and reverse chemogenomics benefit from unified platforms that seamlessly integrate diverse data types. These platforms typically feature:
- Integrated Data Pipelines: Tools like MolPipeline and KNIME provide workflow environments for combining cheminformatic and bioinformatic processing steps [59].
- Heterogeneous Graph Databases: Network structures that connect compounds, targets, pathways, and phenotypes in a unified knowledge graph, enabling complex relationship queries [59].
- Multi-scale Modeling Environments: Platforms that incorporate molecular-level interactions with cellular and pathway-level consequences, bridging chemical and biological scales [63].
Table 2: Methodological Comparison of Forward and Reverse Chemogenomics
| Aspect | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotypic observation [1] [6] | Defined molecular target [1] [6] |
| Primary Screening Method | Phenotypic screening in cells or organisms [35] | Target-based screening (biochemical or virtual) [59] |
| Cheminformatics Focus | Chemical pattern recognition among active compounds [35] | Structure-based design and docking [63] [59] |
| Bioinformatics Focus | Target identification and pathway analysis [1] | Target characterization and family classification [6] |
| Key Data Integration Challenge | Connecting phenotype to molecular target [1] | Connecting target engagement to phenotypic outcome [1] |
| Typical Applications | Mechanism of action studies, phenotypic drug discovery [35] | Rational drug design, target validation [63] |
Essential Research Reagents and Computational Tools
Successful implementation of integrated chemogenomics workflows requires both experimental reagents and computational resources. The following table details key components of the modern chemogenomics toolkit.
Table 3: Research Reagent Solutions and Computational Tools for Integrated Chemogenomics
| Category | Item | Function/Application |
|---|---|---|
| Chemical Libraries | Targeted Chemical Libraries (e.g., kinase-focused, GPCR-focused) | Screening against specific protein families; leveraging similarity principle [1] [6] |
| LOPAC1280 (Library of Pharmacologically Active Compounds) | Reference library for phenotypic screening with annotated activities [6] | |
| Prestwick Chemical Library | Collection of approved drugs for drug repurposing studies [6] | |
| DNA-Encoded Libraries (DELs) | Ultra-large libraries for screening protein-ligand interactions [63] | |
| Bioinformatics Resources | Reference Genomes (hg38) | Standardized reference for genomic alignment and variant calling [62] |
| Protein Data Bank (PDB) | Repository for experimental protein structures [63] | |
| AlphaFold2/AlphaFold3 | AI-based protein structure prediction for targets without experimental structures [63] | |
| Genomic Databases (e.g., gnomAD, COSMIC) | Population variation and cancer mutation data for target prioritization [62] | |
| Computational Tools | RDKit | Open-source cheminformatics platform for molecular descriptor calculation and similarity searching [59] |
| GROMACS | Molecular dynamics simulations for studying protein-ligand interactions [63] | |
| BLAST | Sequence alignment and homology identification [64] | |
| KNIME, Pipeline Pilot | Workflow platforms for building integrated data pipelines [59] | |
| Specialized Assays | Cellular Phenotypic Assays | High-content screening for forward chemogenomics [1] |
| Target-Based Binding Assays | Biochemical screening for reverse chemogenomics (e.g., SPR, FRET) [6] |
Computational Frameworks and Machine Learning Approaches
Advanced computational methods form the backbone of modern data integration strategies in chemogenomics. These approaches enable the prediction of novel drug-target interactions and facilitate the exploration of chemical and biological spaces.
Chemogenomic Machine Learning Models
Machine learning algorithms trained on both chemical and biological data can predict interactions for targets with limited experimental data by leveraging information from related targets and compounds.
- Kronecker Product Methods: These approaches define a joint chemogenomic space using the Kronecker product of protein and ligand descriptors, enabling the prediction of drug-target interactions across entire protein families [35].
- Matrix Factorization: Techniques that decompose the drug-target interaction matrix into lower-dimensional latent representations of both compounds and targets, effectively filling in missing interactions [35].
- Deep Learning Architectures: Multimodal neural networks that process molecular graphs (compounds) and protein sequences simultaneously, learning complex representations that predict binding affinities [6].
These models are particularly valuable for predicting polypharmacology (interactions of compounds with multiple targets) and identifying potential off-target effects early in the drug discovery process [63] [6].
Heterogeneous Knowledge Graphs
Integrated knowledge graphs that connect compounds, targets, diseases, and phenotypes provide a powerful framework for hypothesis generation and drug repurposing.
- Construction: Entities from multiple domains (chemical, biological, clinical) are connected through semantically defined relationships (e.g., "bindsto," "treats," "associatedwith").
- Applications: Graph mining algorithms can identify novel connections between compounds and diseases, suggest new therapeutic uses for existing drugs, and elucidate mechanisms of action [59].
- Examples: The CACTI platform uses clustering analysis to integrate chemogenomic data, revealing patterns and connections within large datasets that might not be apparent through reductionist approaches [59].
Future Directions and Emerging Trends
The field of integrated cheminformatics and bioinformatics continues to evolve rapidly, driven by technological advancements and increasing data availability. Several emerging trends are poised to further transform chemogenomics research:
- AI and Large Language Models: The application of foundation models trained on chemical and biological sequences enables the generation of novel compounds and the prediction of complex interactions [65] [63]. These models can "translate" between chemical structures and biological activities, opening new avenues for drug design.
- Higher-Throughput Free Energy Calculations: Advances in computing hardware and algorithms are making binding affinity calculations through methods like free energy perturbation (FEP) more routine, improving the accuracy of structure-based design [63].
- Multi-omics Integration: The combination of chemogenomic data with other omics technologies (transcriptomics, proteomics, metabolomics) provides systems-level understanding of drug actions [63].
- Quantum Computing: Though still emerging, quantum computing holds promise for accurately simulating molecular interactions and properties that are computationally intractable with classical computers [61].
Effective integration of cheminformatics and bioinformatics data has become indispensable for advancing chemogenomics research. The distinct yet complementary paradigms of forward and reverse chemogenomics require tailored data integration strategies—whether beginning with phenotypic observations and progressing toward target identification, or starting with defined molecular targets and elucidating phenotypic consequences.
The practices outlined in this guide—standardized data representation, unified computational platforms, machine learning approaches, and appropriate reagent selection—provide a framework for maximizing the synergies between chemical and biological data domains. As the volume and complexity of data continue to grow, and as new computational technologies emerge, these integration practices will play an increasingly critical role in accelerating drug discovery and improving our understanding of biological systems.
By adopting these best practices, researchers can more effectively navigate the complex landscape of drug-target interactions, ultimately leading to more efficient identification of novel therapeutic agents and better understanding of their mechanisms of action.
Strategic Decision-Making: A Direct Comparison and Future Outlook
Chemogenomics represents a systematic framework for investigating biological systems and discovering new drugs by screening targeted chemical libraries against entire families of proteins [6] [1]. This field operates on the fundamental principle that studying all possible drug-target interactions across the proteome can accelerate both target validation and compound discovery [1]. Within this domain, two distinct experimental paradigms have emerged: forward chemogenomics and reverse chemogenomics [6] [1]. These approaches differ fundamentally in their starting points and strategic directions, with forward chemogenomics beginning with a phenotypic observation and reverse chemogenomics initiating from a specific protein target [1]. This analysis provides a comprehensive technical comparison of these methodologies, examining their respective strengths, weaknesses, and applications within modern drug discovery pipelines.
Core Conceptual Frameworks
Forward Chemogenomics
Forward chemogenomics, also termed "classical chemogenomics," employs a phenotype-first strategy [1]. Researchers begin by identifying small molecules that induce a specific phenotypic response in cells or whole organisms without prior knowledge of the molecular mechanism involved [6] [1]. The core objective is to use these bioactive compounds as tools to identify the protein targets responsible for the observed phenotype [1]. This approach is particularly valuable for investigating biological pathways where the key molecular players are unknown, allowing the discovery of novel drug targets based on functional outcomes [6].
Reverse Chemogenomics
Reverse chemogenomics adopts a target-first strategy, beginning with a specific protein target and seeking compounds that modulate its activity [6] [1]. This approach initially identifies small molecules that perturb the function of a defined enzyme or receptor in simplified in vitro systems [1]. Once modulators are identified, researchers then analyze the phenotypic consequences of target modulation in cellular or whole-organism contexts [1]. This methodology closely resembles traditional target-based drug discovery but is enhanced by parallel screening capabilities across multiple targets within the same protein family [6] [1].
The conceptual relationship and workflow between these approaches are illustrated below:
Diagram 1: Forward vs. Reverse Chemogenomics Workflows. Forward chemogenomics (red) begins with phenotypic observation, while reverse chemogenomics (blue) initiates from a defined protein target. The approaches can inform one another cyclically.
Comprehensive Comparative Analysis
The following table provides a detailed technical comparison of the core characteristics, strengths, and limitations of forward versus reverse chemogenomics approaches:
Table 1: Head-to-Head Comparison of Forward and Reverse Chemogenomics
| Parameter | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Fundamental Strategy | Phenotype-first approach; begins with observed cellular/organismal phenotype [1] | Target-first approach; begins with predefined protein target [1] |
| Primary Screening Method | Phenotypic screening in biologically relevant systems (cells, tissues, organisms) [45] | Target-based screening using defined in vitro assays (enzymatic, binding) [1] |
| Target Identification | Post-screening target deconvolution required; often challenging [6] [1] | Target known prior to screening; no deconvolution needed [1] |
| Key Strengths | • Discovers novel biology and unexpected targets• Identifies first-in-class therapies with novel mechanisms [45]• Accounts for cellular context and bioavailability [6] | • Streamlined, target-focused process• Enables parallel screening across target families [6] [1]• Straightforward structure-activity relationship (SAR) development [6] |
| Major Limitations | • Target deconvolution is complex and often unsuccessful [6] [1]• Phenotypic assays may have lower throughput [45] | • Limited to known, druggable targets [45]• May miss relevant biology outside predefined target [45]• Compounds may lack cellular activity despite in vitro efficacy [1] |
| Chemical Library Requirements | Diverse compound libraries covering broad chemical space; biologically annotated collections preferred [45] | Targeted libraries focused on specific protein families (kinases, GPCRs, etc.); chemogenomic sets [6] [1] |
| Hit Validation Complexity | High; requires extensive follow-up studies to establish mechanism of action (MOA) [6] [1] | Moderate; focused on confirming on-target activity in cellular contexts [1] |
| Therapeutic Area Fit | Ideal for complex diseases with poorly understood mechanisms [45] | Suitable for well-validated targets with established biology [45] |
| Success Examples | PARP inhibitors for BRCA-mutant cancers, lumacaftor (cystic fibrosis), risdiplam (spinal muscular atrophy) [45] | Most targeted therapies (kinase inhibitors, receptor modulators) [6] |
Experimental Methodologies and Protocols
Forward Chemogenomics Workflow
Protocol 1: Phenotype-Driven Target Discovery
Phenotypic Assay Development: Establish a robust, biologically relevant assay measuring a disease-related phenotype (e.g., tumor cell death, neurite outgrowth, viral infection) [45]. Implement appropriate controls and validation experiments to ensure assay specificity and reproducibility.
Compound Library Screening: Screen diverse chemical libraries, typically comprising 10,000-100,000 compounds, using the phenotypic assay [45]. Prioritize libraries with known bioactivity annotations (e.g., LOPAC1280, Prestwick Chemical Library) to facilitate subsequent target deconvolution [6] [45].
Hit Confirmation and Characterization: Confirm primary hits through dose-response experiments (EC50 determination) and counter-screens to exclude assay artifacts [45]. Assess compound toxicity and specificity within the phenotypic context.
Target Deconvolution - Experimental Approaches:
- Chemical Proteomics: Use immobilized compound analogs to capture and identify interacting proteins from cell lysates [6].
- Transcriptomic Profiling: Compare gene expression signatures of active compounds to reference databases (e.g., Connectivity Map) to infer mechanism [45].
- Resistance Mutagenesis: Generate resistant cell lines and identify mutations through whole-exome sequencing to reveal potential targets [45].
- Biochemical Fractionation: Separate cellular components and track activity through purification to identify target proteins [6].
Target Validation: Confirm target engagement using cellular thermal shift assays (CETSA), biophysical methods, and genetic approaches (CRISPR, RNAi) to establish causal relationship between target modulation and phenotype [6] [45].
The following diagram illustrates the experimental decision points in selecting the appropriate target deconvolution method:
Diagram 2: Target Deconvolution Strategy Map for Forward Chemogenomics. This decision tree guides selection of appropriate experimental methods based on compound characteristics and available resources.
Reverse Chemogenomics Workflow
Protocol 2: Target-Centric Ligand Discovery
Target Selection and Validation: Select a biologically validated protein target from a therapeutically relevant family (e.g., kinases, GPCRs, nuclear receptors) [6] [1]. Confirm target relevance to disease pathophysiology through genetic and clinical evidence.
Assay Development: Establish robust in vitro assays measuring target modulation (e.g., enzymatic activity, receptor binding, protein-protein interaction) [1]. Implement appropriate counter-screens to identify promiscuous inhibitors or assay interferants.
Focused Library Screening: Screen targeted chemogenomic libraries specifically designed for the target family of interest [6] [1]. These libraries typically contain compounds with known activity against related targets, leveraging family-wide structural similarities [6].
Hit-to-Lead Optimization:
- Selectivity Profiling: Screen confirmed hits against related targets within the same family to establish selectivity profiles [6] [66].
- Structure-Activity Relationship (SAR): Systematically modify hit compounds to improve potency, selectivity, and physicochemical properties [6].
- Structural Biology: Utilize protein-ligand co-crystal structures to guide rational design of improved compounds [6].
Cellular Target Engagement: Confirm compound activity in cellular contexts using pharmacodynamic assays measuring downstream pathway modulation [1] [66].
Phenotypic Validation: Test optimized compounds in disease-relevant phenotypic assays to confirm therapeutic hypothesis and identify potential polypharmacology [1].
Research Reagents and Essential Materials
Successful implementation of chemogenomics approaches requires carefully selected reagent systems and compound libraries. The following table details key research tools essential for both forward and reverse chemogenomics studies:
Table 2: Essential Research Reagents for Chemogenomics Studies
| Reagent Category | Specific Examples | Function and Application | Suitability |
|---|---|---|---|
| Chemogenomic Compound Libraries | EUbOPEN Library [67], GSK Biologically Diverse Compound Set [6], Pfizer Chemogenomic Library [6] | Targeted collections covering specific protein families; enable systematic exploration of target space [6] [67] | Both approaches |
| Annotated Bioactive Collections | LOPAC1280 [6], Prestwick Chemical Library [6], NCATS Mechanism Interrogation PlatE 3.0 [6] | Libraries with known mechanism-of-action; facilitate target hypothesis generation and deconvolution [6] [45] | Primarily forward |
| Cell-Based Assay Systems | Primary patient-derived cells [67], iPSC-derived models [45], 3D organoids [68] | Biologically relevant systems for phenotypic screening; improve translational predictivity [45] [68] | Primarily forward |
| Protein Production Systems | Recombinant protein expression (E. coli, insect, mammalian cells) [66] | Production of purified, functional protein targets for in vitro screening [1] | Primarily reverse |
| Target Engagement Assays | Cellular Thermal Shift Assay (CETSA) [66], Bioluminescence Resonance Energy Transfer (BRET) [6] | Confirm compound binding to intended targets in physiological environments [6] [66] | Both approaches |
| Multi-omics Readouts | RNA sequencing, Proteomics, Metabolomics platforms [45] | Comprehensive molecular profiling for mechanism elucidation and biomarker identification [45] | Both approaches |
Integrated Applications and Case Studies
Case Study 1: NR4A Nuclear Receptor Ligand Discovery
A recent investigation demonstrated the power of reverse chemogenomics for challenging target families [66]. Researchers systematically profiled reported NR4A nuclear receptor modulators using orthogonal cellular and biophysical assays, validating a set of eight high-quality chemical tools from initially promising compounds [66]. This curated chemogenomic set enabled exploration of NR4A biology in endoplasmic reticulum stress and adipocyte differentiation, demonstrating the utility of well-characterized compound sets for target validation [66].
Case Study 2: EUbOPEN Consortium - Systematic Target Coverage
The EUbOPEN consortium represents a large-scale implementation of chemogenomics principles, developing open-access chemical tools for biological exploration and target validation [67]. This public-private partnership has created a chemogenomic library covering approximately one-third of the druggable proteome, along with hundreds of high-quality chemical probes [67]. This systematic approach enables both forward screening campaigns and reverse target validation studies across multiple target families.
Integration with Artificial Intelligence and Machine Learning
Modern chemogenomics increasingly leverages machine learning (ML) and artificial intelligence (AI) to overcome traditional limitations [69]. For forward chemogenomics, ML models can predict targets based on chemical structure and phenotypic profiles, accelerating target deconvolution [69]. In reverse chemogenomics, deep learning approaches enable prediction of drug-target interactions across entire proteomes, facilitating polypharmacology profiling and off-target prediction [6] [69]. Multi-task learning frameworks are particularly valuable for predicting activity across multiple targets simultaneously, supporting rational polypharmacology design [69].
Forward and reverse chemogenomics represent complementary rather than competing strategies in modern drug discovery. The optimal approach depends on the specific biological question, available tools, and stage of therapeutic development. Forward chemogenomics excels at novel biology and target discovery, while reverse chemogenomics provides a streamlined path for validated targets. The most successful drug discovery programs increasingly integrate elements of both approaches, using phenotypic screening to identify novel biology and target-centric methods to optimize chemical tools.
Future developments will likely focus on overcoming the key limitations of both approaches. For forward chemogenomics, improved target deconvolution technologies represent a critical need, with emerging methods in chemical proteomics, functional genomics, and artificial intelligence showing particular promise [69]. For reverse chemogenomics, expanding the druggable proteome beyond traditional target families remains a priority, with initiatives like Target 2035 aiming to develop chemical probes for most human proteins by 2035 [67]. The continued integration of chemogenomics with systems pharmacology, multi-omics technologies, and machine learning will further enhance our ability to discover and develop novel therapeutics for complex diseases.
Chemogenomics represents a systematic approach to drug discovery that screens small molecule libraries against families of drug targets to identify novel drugs and targets [1]. This field operates through two primary, complementary paradigms: forward chemogenomics and reverse chemogenomics.
In forward chemogenomics (also termed classical chemogenomics), research begins with the observation of a desired biological phenotype, such as the arrest of tumor growth. The objective is to identify small molecules that induce this phenotype and then use these molecules as tools to discover the specific protein target responsible for the observed effect [1]. This is a "phenotype-first" approach.
Conversely, reverse chemogenomics starts with a defined protein target of interest, such as a specific enzyme. Researchers first identify small molecules that perturb the target's function in an in vitro assay. These modulators are then analyzed in cellular or whole-organism models to understand the resulting phenotype and confirm the target's biological role [1]. This strategy aligns closely with traditional target-based drug discovery but is enhanced by parallel screening across entire target families.
The following diagram illustrates the core logical workflow of each approach, highlighting their distinct starting points and experimental trajectories.
Choosing the correct path is critical for project success, as the decision influences experimental design, resource allocation, and the interpretation of results. This guide provides a structured framework for making that choice.
Decision Framework: Forward vs. Reverse Chemogenomics
The choice between forward and reverse chemogenomics is multifaceted. The table below summarizes the key project characteristics that should guide this strategic decision.
Table 1: Project Characteristics and Recommended Chemogenomics Approaches
| Project Characteristic | Recommended Approach | Rationale |
|---|---|---|
| Starting Point | Unknown molecular mechanism; complex phenotype | Defined protein target with known/predicted function |
| Primary Goal | Discover novel targets & mechanisms | Validate a target's role in biology; optimize known binders |
| Phenotypic Assay | Available, robust, & disease-relevant | Not required for initial screening; used later for validation |
| Target Family Knowledge | Limited; exploring orphan targets | Substantial; leveraging known ligands & SAR |
| Risk Tolerance | Higher risk, potential for breakthrough findings | Lower risk, more predictable and focused path |
| Key Strength | Unbiased discovery of novel biology [3] | High efficiency for lead optimization across target families [1] |
| Major Challenge | Deconvoluting target from phenotype [1] | May overlook complex biology or off-target effects |
Experimental Protocols for Forward and Reverse Chemogenomics
Forward Chemogenomics Workflow
The forward approach requires a robust phenotypic screen followed by a often complex target identification phase.
Phase 1: Phenotypic Screening
- Assay Development: Establish a high-content or high-throughput phenotypic assay that reliably measures a disease-relevant endpoint (e.g., cell viability, morphological change, reporter gene expression) [3].
- Library Screening: Screen a diverse, drug-like chemical library against the assay system. Using libraries with "privileged structures," such as those derived from traditional medicines, can be advantageous due to their favorable solubility and safety profiles [1].
- Hit Confirmation: Identify and confirm "hits" – compounds that reproducibly induce the desired phenotype. Secondary assays should be used to rule out false positives.
Phase 2: Target Deconvolution This is the most critical and challenging phase. Key methodologies include:
- Chemical-Genetic Interaction Profiling (CGIP): This powerful method involves systematically measuring how a drug affects the fitness of a genome-wide collection of mutant strains (e.g., gene knockouts or knockdowns) [70] [19].
- Pooled Fitness Screens: Barcoded mutant libraries (e.g., yeast knockout collection) are grown competitively in the presence of the hit compound. Sequencing the barcodes before and after treatment reveals which gene deletions make cells more sensitive or resistant to the drug, directly implicating those genes in the compound's mechanism of action [19].
- Haploinsufficiency Profiling (HIP): Used in diploid organisms, HIP screens a library of strains where one copy of an essential gene is deleted. If the drug target is an essential gene, reducing its gene dosage (haploinsufficiency) often makes the cell hypersensitive to the drug, helping to identify the target [70] [19].
- Affinity Purification: Chemically modify the hit compound to create an affinity matrix. Incubate this with cell lysates, pull down bound proteins, and identify them via mass spectrometry.
- Transcriptomic Profiling: Compare the global gene expression signature induced by the hit compound to a database of signatures from compounds with known mechanisms of action (guilt-by-association) to infer its mode of action [19].
Reverse Chemogenomics Workflow
The reverse approach is more linear, beginning with a specific protein target.
Phase 1: Target-Based Screening
- Target Selection & Production: Select a purified protein target (e.g., a kinase, GPCR, or protease) and develop a high-throughput in vitro biochemical assay to measure its activity (e.g., enzymatic rate, ligand binding) [1] [6].
- Focused Library Screening: Screen a targeted chemical library. These libraries are often enriched with known ligands for other members of the same target family, increasing the probability of finding hits due to structural similarities within the family [1].
- Hit Identification: Identify compounds that show potent activity in the in vitro assay (e.g., inhibition or activation).
Phase 2: Phenotypic Validation
- Cellular Assay Testing: Test the confirmed hits in a cellular model to determine if the in vitro activity translates to a functional effect in a more complex biological system (e.g., inhibiting cell signaling, changing metabolite levels).
- Mechanism of Action (MoA) Studies: Use techniques such as Target Overexpression: If overexpressing the target protein in cells makes them resistant to the compound, it provides strong evidence that the protein is the compound's primary target in vivo [70].
- Lead Optimization: Use the initial hit as a starting point for medicinal chemistry to improve its potency, selectivity, and drug-like properties, often screening against related targets to minimize off-target effects [1].
The following workflow diagram encapsulates the key stages and decision points in both the forward and reverse chemogenomics pathways.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of chemogenomic strategies relies on specialized biological and chemical tools. The following table details key resources used in the featured experiments.
Table 2: Key Research Reagent Solutions for Chemogenomics
| Reagent / Resource | Function and Application | Key Characteristics |
|---|---|---|
| Barcoded Mutant Libraries (e.g., Yeast KO collection) [19] | Enables competitive fitness profiling in pooled screens. Essential for identifying chemical-genetic interactions in forward chemogenomics. | Each strain has unique DNA barcodes; allows parallel fitness measurement via sequencing. |
| Targeted Chemical Libraries (e.g., Kinase-focused, GPCR-focused) [1] [6] | Used in reverse chemogenomics to screen specific target families. Increases hit rate by leveraging known ligand chemotypes. | Contains known ligands for at least one family member; designed for high "hit-rate". |
| Haploinsufficiency (HIP) & Homozygous Profiling (HOP) Libraries [70] [19] | HIP: Identifies drug targets (essential genes). HOP: Reveates resistance/sensitivity mechanisms (non-essential genes). | HIP has one essential gene copy deleted; HOP has non-essential genes fully deleted. |
| CRISPRi/a Knockdown/Activation Libraries [70] | Enables targeted gene knockdown (CRISPRi) or activation (CRISPRa) in mammalian cells for chemical-genetic screens. | Genome-wide; allows modulation of gene dosage in human cell lines for MoA studies. |
| Phenotypic Assay Kits (e.g., Cell Painting, High-Content Imaging) [3] | Provides multi-parametric profiling of cell morphology in response to compounds. Used for phenotypic screening and MoA classification. | Uses fluorescent dyes to mark organelles; generates rich, high-dimensional data. |
| DNA-Encoded Chemical Libraries (DEL) | Allows screening of ultra-large compound libraries (billions of members) by tagging each molecule with a DNA barcode. | Extremely large library size; identification of binders via affinity selection and DNA sequencing. |
The strategic selection between forward and reverse chemogenomics is a foundational decision that sets the trajectory for a drug discovery project. Forward chemogenomics offers an unbiased path to novel target discovery when beginning with a robust phenotype but carries the challenge of subsequent target deconvolution. Reverse chemogenomics provides a focused and efficient route for validating targets and optimizing leads when a hypothesis about a specific protein's role exists. The most modern drug discovery pipelines are increasingly hybrid, leveraging the unbiased power of phenotypic screening (forward) and then using advanced chemical-genetic and computational tools for rapid target identification (a reverse principle) to create an integrated, iterative, and powerful strategy for delivering new therapeutics.
The paradigm of drug discovery has long been divided between target-based and phenotypic approaches. Target-based drug discovery (TDD) relies on a established causal relationship between a specific molecular target and a disease, whereas phenotypic drug discovery (PDD) focuses on modulating a disease phenotype or biomarker without a pre-specified target hypothesis [71]. This dichotomy provides the foundation for understanding the two principal strategies in chemogenomics: forward and reverse.
Forward chemogenomics (often phenotype-based) starts with a biological phenotype of interest and employs chemical tools as probes to identify the protein targets responsible for the observed phenotypic effect. Conversely, reverse chemogenomics (often target-based) begins with a defined molecular target and uses chemical ligands to elucidate its biological function and therapeutic potential [72] [11]. The validation framework connecting these approaches ensures that chemical probes not only engage their intended targets but also elicit biologically relevant phenotypic outcomes, creating a crucial bridge between molecular and phenotypic understanding.
This technical guide outlines comprehensive validation frameworks for assessing target engagement and phenotypic relevance within this integrated chemogenomics paradigm, providing methodologies and tools for researchers navigating the complex journey from chemical hit to validated therapeutic candidate.
Target Engagement Assessment Frameworks
Target engagement (TE) validation confirms that a compound physically interacts with its intended macromolecular target in a physiologically relevant context. This requires multiple orthogonal methods to provide compelling evidence for specific binding.
Biochemical and Biophysical Assays
Direct binding assays form the foundation of TE assessment, providing quantitative parameters about the compound-target interaction.
Table 1: Biochemical and Biophysical TE Assessment Methods
| Method | Measured Parameters | Throughput | Key Applications |
|---|---|---|---|
| Surface Plasmon Resonance (SPR) | Binding kinetics (kon, koff), affinity (KD) | Medium | Direct label-free binding measurement in real-time |
| Isothermal Titration Calorimetry (ITC) | Binding affinity (KD), stoichiometry (n), enthalpy (ΔH), entropy (ΔS) | Low | Thermodynamic characterization of binding interactions |
| Cellular Thermal Shift Assay (CETSA) | Thermal stabilization, apparent affinity | Medium-high | Intracellular TE, membrane permeability assessment |
| Bioluminescence Resonance Energy Transfer (BRET) | Proximity, binding events in live cells | High | Intracellular TE, kinetic monitoring in physiological environments |
Experimental Protocols for Key TE Assays
Cellular Thermal Shift Assay (CETSA) Protocol
The CETSA method evaluates target engagement in intact cellular environments by detecting ligand-induced thermal stabilization of target proteins.
- Cell Preparation and Treatment: Culture cells expressing the target protein of interest in appropriate medium. Harvest cells at 70-80% confluence and prepare single-cell suspensions. Treat cell aliquots with compound of interest or vehicle control (typically DMSO) for predetermined time (e.g., 1-4 hours) at physiologically relevant concentrations.
- Heat Challenge: Divide compound-treated and control cell suspensions into smaller aliquots (typically 50-100 μL). Subject each aliquot to different precisely controlled temperatures (e.g., range from 37°C to 65°C) for 3-5 minutes using a thermal cycler or precise water bath.
- Cell Lysis and Protein Separation: Following heat challenge, freeze-thaw cycles or chemical lysis to disrupt cell membranes. Separate soluble protein fraction from precipitated material by high-speed centrifugation (20,000 × g for 20 minutes at 4°C).
- Target Protein Detection: Analyze soluble protein fractions by Western blotting or quantitative mass spectrometry to determine amount of target protein remaining soluble at each temperature.
- Data Analysis: Calculate melting temperature (Tm) shifts between compound-treated and vehicle control samples. Significant positive Tm shifts (typically >2°C) indicate stabilization due to compound binding and successful target engagement.
Bioluminescence Resonance Energy Transfer (BRET) TE Protocol
BRET enables real-time monitoring of target engagement in live cells under physiological conditions.
- Construct Design: Create fusion proteins of target of interest with either BRET donor (e.g., NanoLuc luciferase) or acceptor (e.g., fluorescent protein HaloTag). For competitive binding assays, fuse donor to target protein and label acceptor with cell-permeable fluorescent ligand.
- Cell Transfection and Plating: Transfect appropriate host cells (HEK293T, CHO, etc.) with donor and acceptor constructs. Plate transfected cells in white-walled, clear-bottom 96- or 384-well plates suitable for luminescence detection.
- Acceptor Labeling: Incubate cells with cell-permeable HaloTag ligand conjugated to BRET-acceptor fluorophore (e.g., 50-100 nM for 30-60 minutes). Wash cells to remove excess ligand.
- Compound Treatment and BRET Measurement: Treat cells with test compounds at various concentrations. Add cell-permeable luciferase substrate (e.g., furimazine). Measure both donor emission (450-470 nm) and acceptor emission (550-600 nm) using a plate reader capable of simultaneous luminescence/fluorescence detection.
- Data Analysis: Calculate BRET ratio as (acceptor emission)/(donor emission). Plot concentration-response curves to determine IC50 values for competitive displacement. Reduced BRET signal indicates compound binding and displacement of the fluorescent tracer, confirming target engagement.
Phenotypic Relevance Validation
Phenotypic relevance validation ensures that target engagement translates to meaningful biological outcomes in physiologically relevant models. This is particularly critical in forward chemogenomics approaches where the molecular target may be unknown initially.
Phenotypic Screening Hit Triage and Validation
Successful hit validation in phenotypic screening relies on three types of biological knowledge: known mechanisms, disease biology, and safety considerations [73]. Structure-based hit triage alone may be counterproductive in phenotypic approaches, as the most promising hits may act through novel mechanisms of action.
Table 2: Phenotypic Validation Assays Across Biological Complexity
| Complexity Level | Assay Types | Readouts | Validation Strengths |
|---|---|---|---|
| Pathway/Network | Reporter gene assays, pathway enrichment, phospho-flow cytometry | Transcriptional activation, phosphorylation status, second messenger levels | Mechanism deconvolution, network biology understanding |
| Cellular | High-content imaging, 2D/3D proliferation, cytotoxicity, migration | Morphological changes, viability, motility, synaptic activity | Contextual biology, functional outcomes in relevant cell types |
| Tissue/Organoid | Patient-derived organoids, tissue explants, precision-cut slices | Architecture preservation, multicellular interactions, tissue-level functions | Microphysiological systems, human disease relevance |
| Whole Organism | Zebrafish, rodent disease models, phenotypic rescue | Disease modification, behavioral improvement, survival extension | Systems-level integration, ADME/PK considerations |
Experimental Protocols for Phenotypic Validation
High-Content Imaging Phenotypic Profiling Protocol
Multi-parameter high-content imaging enables quantitative assessment of phenotypic changes in relevant cellular models.
- Cell Model Selection and Plating: Select disease-relevant cell types (primary cells, iPSC-derived cells, or engineered cell lines). Plate cells in optical-grade 96- or 384-well plates at optimized density for the specific assay duration. Include appropriate controls (vehicle, positive control compounds, negative controls).
- Compound Treatment and Staining: Treat cells with test compounds across a concentration range (typically 8-point dilution series) for biologically relevant timeframes (24-72 hours). Fix cells with paraformaldehyde (4% for 15 minutes) and permeabilize with Triton X-100 (0.1-0.5% for 10 minutes). Block with appropriate serum (5-10% for 1 hour). Incubate with validated primary antibodies targeting phenotypic markers of interest (e.g., cytoskeletal proteins, organelle markers, signaling proteins) followed by fluorescently-labeled secondary antibodies and counterstains (e.g., DAPI for nuclei, phalloidin for actin).
- Image Acquisition: Acquire images using high-content imaging system (e.g., ImageXpress, Operetta, or CellInsight) with appropriate objectives (20x or 40x) at multiple sites per well (minimum 4-9 sites) to ensure statistical robustness. Maintain identical acquisition settings across all experimental conditions.
- Image Analysis and Feature Extraction: Use integrated software (e.g., CellProfiler, Harmony, or custom pipelines) to segment cells and subcellular compartments. Extract morphological features (size, shape, texture), intensity features (expression levels, localization), and contextual features (neighbor interactions, spatial patterns). Typically extract 500-1000 features per cell.
- Multivariate Analysis and Phenotypic Scoring: Apply dimensionality reduction techniques (PCA, t-SNE) and clustering algorithms to identify distinct phenotypic profiles. Use machine learning approaches to classify compound treatments based on their phenotypic fingerprints. Compare to reference compounds with known mechanisms to generate hypotheses about potential mechanisms of action.
Phenotypic Rescue in Complex Disease Models Protocol
Demonstrating dose-dependent reversal of disease phenotypes in physiologically relevant models provides compelling evidence for phenotypic relevance.
- Model Establishment: Select appropriate disease model based on biological question: patient-derived organoids, 3D spheroid cultures, or whole organism models (zebrafish, Drosophila, rodent). For cellular models, establish robust phenotypic readouts of disease state (e.g., protein aggregation for neurodegenerative diseases, electrophysiological parameters for channelopathies, contraction force for cardiac diseases).
- Baseline Phenotype Characterization: Quantitatively characterize the disease phenotype in untreated models using established assays. For genetic models, confirm genotype-phenotype correlation. Include appropriate controls (wild-type, disease controls, vehicle-treated controls).
- Compound Treatment and Phenotypic Assessment: Treat models with test compounds across a concentration range. Include positive control compounds with known efficacy if available. Assess phenotypic endpoints at appropriate timepoints post-treatment. For chronic models, consider multiple timepoints to assess durability of effect.
- Dose-Response Analysis: Fit concentration-response curves to determine EC50 values for phenotypic rescue. Evaluate maximum efficacy (% reversal of disease phenotype) compared to positive controls and normal baseline.
- Specificity Assessment: Conduct counter-screens against related but distinct phenotypes to evaluate specificity of effect. Use orthogonal assays to confirm phenotypic rescue through different readout modalities.
Integrating Forward and Reverse Chemogenomics Approaches
The power of modern validation frameworks lies in their ability to bridge forward and reverse chemogenomics approaches, creating an iterative cycle of hypothesis generation and testing.
Mechanism of Action Deconvolution
For hits identified in phenotypic screens (forward chemogenomics), mechanism of action deconvolution is essential for target identification and validation.
Quantitative Framework for Integrated Validation
Establishing quantitative relationships between target engagement, pathway modulation, and phenotypic response creates a robust validation framework.
Table 3: Quantitative Parameters for Integrated Validation
| Validation Tier | Key Parameters | Acceptance Criteria | Experimental Approaches |
|---|---|---|---|
| Target Engagement | Cellular IC50/EC50, Residence time, Occupancy-efficacy relationship | >50% target engagement at efficacious concentrations, sustained engagement | CETSA, BRET, PET imaging, occupancy assays |
| Pathway Modulation | Pathway EC50, Modulation magnitude, Onset/offset kinetics | Pathway modulation precedes phenotypic effect, maximal pathway engagement | Phospho-flow, reporter genes, transcriptional profiling |
| Phenotypic Response | Phenotypic EC50, Maximal efficacy, Therapeutic index | Dose-dependent response, efficacy comparable to standards | High-content imaging, functional assays, disease models |
| Translational Concordance | Species differences, Biomarker correlation, Clinical translatability | Conservation across species, biomarker confirmation | Cross-species testing, biomarker development |
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of validation frameworks requires carefully selected research tools and reagents. The following table outlines essential solutions for comprehensive target engagement and phenotypic assessment.
Table 4: Essential Research Reagent Solutions for Validation Studies
| Reagent Category | Specific Examples | Key Functions | Application Notes |
|---|---|---|---|
| Tagged Protein Systems | HaloTag, SNAP-tag, HALO-/-NanoLuc fusions | Protein labeling, pulse-chase experiments, fusion protein construction | Enable specific labeling with fluorescent or biotinylated ligands for tracking and engagement studies |
| Cellular Dielectric Spectroscopy | CellKey, xCELLigence systems | Label-free cellular response profiling, real-time functional assessment | Measure impedance changes for kinetic response assessment without labels |
| Biosensor Platforms | EPAC cAMP biosensors, kinase translocation reporters | Second messenger detection, pathway activation monitoring | Live-cell monitoring of pathway modulation with temporal resolution |
| Chemical Proteomics Kits | ActivX probes, kinobeads, photoaffinity labeling kits | Target identification, selectivity profiling, engagement assessment | Covalent modification of target families for pull-down and identification |
| Genome Editing Tools | CRISPR/Cas9 systems, RNAi libraries | Target validation, genetic dependency assessment | Knockout/knockdown studies to confirm target necessity for phenotype |
| Advanced Cell Models | iPSC-derived cells, patient-derived organoids, 3D spheroids | Disease modeling, physiological relevance | Human genetic context preservation, complex phenotypic assessment |
| Multiplexed Assay Reagents | Luminex kits, MSD panels, LEGENDplex arrays | Multi-analyte profiling, cytokine/phosphoprotein measurement | Simultaneous measurement of multiple analytes from limited samples |
Robust validation frameworks that simultaneously assess target engagement and phenotypic relevance are essential for successful drug discovery in both forward and reverse chemogenomics paradigms. The integrated approaches outlined in this guide provide a comprehensive pathway for establishing confidence in compound mechanism and therapeutic potential. By quantitatively linking molecular interactions to phenotypic outcomes across multiple layers of biological complexity, researchers can derisk therapeutic candidates and prioritize those with the highest probability of clinical success. As chemogenomics continues to evolve, these validation frameworks will increasingly incorporate computational approaches, machine learning, and multi-omics data integration to further enhance predictive power and translation to human disease.
The Growing Role in Drug Repurposing and Polypharmacology
Drug repurposing, the systematic identification of new therapeutic indications for existing drugs, represents a paradigm shift in pharmaceutical research by offering reduced development timelines, lower costs, and decreased failure rates compared to traditional drug discovery [74]. This approach has gained significant traction within the broader framework of chemogenomics, which explores the systematic relationship between chemical compounds and biological targets across genomic space. Within chemogenomics, two distinct research strategies have emerged: forward chemogenomics and reverse chemogenomics [19].
Forward chemogenomics begins with a biological perturbation—such as a gene deletion or overexpression—and assesses the effects of chemical compounds on the resulting phenotype. This approach is particularly valuable for identifying mechanisms of drug action (MODA) when a compound produces a phenotype of interest but its target remains unknown. In contrast, reverse chemogenomics starts with a specific protein target of interest and screens for compounds that modulate its activity [19]. Both paradigms generate rich chemogenomic profiles that serve as valuable resources for drug repurposing and polypharmacology—the study of how single drugs can interact with multiple targets to produce complex therapeutic effects.
Artificial intelligence (AI) has dramatically accelerated both forward and reverse chemogenomics approaches by enabling the analysis of complex, high-dimensional datasets that would be intractable through manual methods [74]. AI-driven techniques can identify non-obvious drug-disease associations and polypharmacological relationships by integrating diverse data sources including chemical structures, protein sequences, interaction networks, and clinical profiles [44]. This technical guide explores the computational frameworks, experimental methodologies, and data resources that underpin modern drug repurposing efforts within forward and reverse chemogenomics paradigms.
Computational Frameworks for Repurposing
Pattern Recognition and Machine Learning Approaches
Pattern recognition algorithms have become indispensable tools for analyzing chemogenomic data in drug repurposing. These approaches can be broadly categorized into traditional machine learning and deep learning techniques, each with distinct strengths for extracting patterns from different data types.
Traditional machine learning algorithms applied in pharmacogenomics and repurposing include:
- Support Vector Machines (SVM): Used for classification tasks such as predicting treatment responders vs. non-responders based on genetic profiles [44]
- Random Forests (RF): Employed for feature selection and identifying predictive genetic markers from high-dimensional data [44]
- Logistic Regression: Applied to model binary outcomes in drug response [74]
Deep learning architectures offer enhanced capability for capturing complex, non-linear relationships in large-scale datasets:
- Multilayer Perceptron (MLP): A feed-forward artificial neural network suitable for data escalation [74]
- Convolutional Neural Networks (CNN): Ideal for processing structural and sequence data [4]
- Long Short-Term Memory Recurrent Neural Networks (LSTM-RNN): Compatible with sequential data analysis [74]
- Graph Neural Networks (GNN): Effective for representing molecular structures as graphs [4]
Table 1: Machine Learning Approaches in Drug Repurposing
| Algorithm Type | Representative Models | Primary Applications in Repurposing | Data Requirements |
|---|---|---|---|
| Traditional ML | SVM, Random Forests, Logistic Regression | Drug-response classification, biomarker discovery | Structured genomic and clinical data |
| Deep Learning | CNN, LSTM, GNN | Drug-target affinity prediction, molecular generation | Raw sequence, structural, and interaction data |
| Multitask Learning | DeepDTAGen | Simultaneous affinity prediction and drug generation | Paired drug-target interaction data |
The DeepDTAGen framework exemplifies advanced multitask learning in drug repurposing, which simultaneously predicts drug-target binding affinities and generates novel target-aware drug variants using a shared feature space [4]. This approach addresses a critical challenge in polypharmacology: identifying compounds with specific multi-target profiles. To mitigate optimization challenges such as conflicting gradients between tasks, DeepDTAGen incorporates the FetterGrad algorithm, which maintains gradient alignment by minimizing Euclidean distance between task gradients during model training [4].
Network-Based and Literature-Driven Approaches
Network-based approaches study relationships between molecules—including protein-protein interactions (PPIs), drug-disease associations (DDAs), and drug-target associations (DTAs)—to identify repurposing opportunities based on network proximity [74]. The fundamental premise is that drugs located closer to disease-associated molecular modules in biological networks tend to be more promising repurposing candidates [74]. These methods employ mathematical approaches such as random walks to predict network relationships, where movement between nodes depends on their weight characteristics [74].
Literature-based repurposing represents another powerful approach that leverages the vast corpus of published scientific knowledge. One recent methodology calculated literature-based similarity between drugs using the Jaccard coefficient to measure overlap in their associated research publications [75]. This approach identified 19,553 potential drug pairs for repurposing, with the Jaccard coefficient demonstrating superior performance as a similarity metric compared to other measures [75]. The underlying hypothesis is that drugs sharing substantial literature coverage likely target related biological pathways or processes, suggesting potential for shared therapeutic applications.
Experimental Protocols and Methodologies
Forward Chemogenomics Screening Protocols
Forward chemogenomics approaches are particularly valuable for identifying drug mechanisms when phenotypic screening reveals a compound of interest with unknown molecular targets. The following protocol outlines a comprehensive forward chemogenomics screening methodology:
1. Library Preparation and Screening
- Select compound libraries comprising approved drugs, investigational agents, or diverse chemical collections
- Implement high-throughput or high-content screening against model organisms (e.g., yeast) or cellular systems
- Measure phenotypic endpoints relevant to the disease of interest (e.g., cell viability, morphological changes, reporter gene expression)
2. Chemogenomic Profile Generation
- For yeast systems, utilize the Yeast Knockout (YKO) collection comprising ~6000 deletion strains [19]
- Employ barcoded competitive growth assays in the presence of compounds of interest
- Sequence barcodes to quantify strain abundance and calculate fitness scores for each gene deletion
3. Target Identification and Validation
- Apply guilt-by-association principles by comparing chemogenomic profiles to reference compounds with known mechanisms
- Integrate complementary approaches including HaploInsufficiency Profiling (HIP) and Homozygous Profiling (HOP) to enhance target identification [19]
- Validate putative targets through secondary assays including biochemical binding studies and genetic rescue experiments
4. Data Integration and Repurposing Hypothesis Generation
- Integrate chemogenomic profiles with additional functional genomics data (e.g., transcriptomic, proteomic)
- Apply pattern recognition algorithms to identify compounds with similar profiles but different indications
- Generate repurposing hypotheses based on shared mechanisms rather than structural similarity
Reverse Chemogenomics and Computational Target Fishing
Reverse chemogenomics begins with a defined molecular target and seeks to identify compounds that modulate its activity. The following protocol describes a computational target fishing approach for drug repurposing:
1. Target Selection and Characterization
- Select therapeutic targets based on genetic evidence, pathway analysis, or disease mechanisms
- Collect structural information (crystal structures, homology models) or sequence data for the target
- Curate known ligands and modulators from databases such as ChEMBL, BindingDB, and GtoPdb [76]
2. Compound Screening and Prioritization
- Screen compound libraries using molecular docking, pharmacophore matching, or similarity searching
- Apply machine learning models (e.g., DeepDTA, GraphDTA) to predict drug-target binding affinities [4]
- Prioritize compounds based on predicted affinity, selectivity, and drug-like properties
3. Multi-Target Profiling and Polypharmacology Assessment
- Evaluate prioritized compounds against secondary targets to identify potential polypharmacology
- Apply network-based methods to assess the therapeutic potential of multi-target profiles
- Use tools such as the repoDB database to identify potential indication expansions [77]
4. Experimental Validation
- Validate computational predictions through in vitro binding assays (e.g., Ki, Kd, IC50 determinations)
- Assess functional activity in cellular models relevant to the new indication
- Evaluate efficacy in animal models of the target disease
Table 2: Key Data Resources for Drug Repurposing
| Resource Name | Data Type | Application in Repurposing | Key Features |
|---|---|---|---|
| ChEMBL | Bioactivity data | Target identification, affinity prediction | 21M+ bioactivity measurements, 16K+ targets [76] |
| BindingDB | Binding affinities | DTA prediction, virtual screening | 2.4M+ binding measurements, ~9K targets [76] |
| GtoPdb | Curated target-ligand interactions | Mechanism-based repurposing | Expert-curated GPCRs, ion channels, nuclear receptors [76] |
| repoDB | Approved/failed drug-indication pairs | Validation and benchmarking | 6,677 approved and 4,123 failed pairs [77] |
| DrugCentral | Drug information | Indication mapping and analysis | UMLS-mapped indications from drug labels [77] |
Successful implementation of drug repurposing strategies requires access to comprehensive data resources, computational tools, and experimental reagents. The following table details essential components of the repurposing toolkit:
Table 3: Research Reagent Solutions for Drug Repurposing
| Resource Category | Specific Tools/Reagents | Function and Application |
|---|---|---|
| Compound Libraries | Approved Drug Libraries, YKO Collection | Screening for phenotypic effects or target identification [19] |
| Bioactivity Databases | ChEMBL, BindingDB, GtoPdb | Source of drug-target interaction data for computational analysis [76] |
| Validation Resources | repoDB, ClinicalTrials.gov | Benchmarking predictions against known successes and failures [77] |
| Computational Frameworks | DeepDTAGen, KronRLS, SimBoost | Predicting drug-target interactions and binding affinities [4] |
| Network Analysis Tools | Cytoscape, NetworkX | Constructing and analyzing drug-target-disease networks [74] |
| Chemical Informatics | RDKit, OpenBabel | Processing chemical structures and calculating molecular descriptors [76] |
Visualization of Repurposing Workflows
Challenges and Future Directions
Despite significant advances, several challenges persist in AI-driven drug repurposing. The translational gap between computational predictions and clinical efficacy remains substantial, as evidenced during the COVID-19 pandemic when many computationally promising candidates failed in clinical trials [44]. This highlights the need for improved model interpretability, better integration of heterogeneous data sources, and more robust validation frameworks.
Additional challenges include:
- Data quality and heterogeneity: Variations in data curation standards, assay protocols, and annotation schemas across sources [76]
- Algorithmic transparency: The "black box" nature of many deep learning models limits mechanistic insights [75]
- Regulatory and financial barriers: Lack of clear pathways for approval and reimbursement of repurposed drugs [78]
Future progress will likely come from enhanced multitask learning frameworks that simultaneously predict drug-target interactions, generate novel compounds, and anticipate adverse effects [4]. Improved knowledge graph embeddings that integrate diverse data types (genomic, clinical, chemical) will enable more comprehensive repurposing hypotheses [44]. Furthermore, collaborative networks such as the UCL Repurposing Therapeutic Innovation Network are emerging to address translational challenges by combining multidisciplinary expertise [78].
The convergence of forward and reverse chemogenomics approaches through AI-driven methodologies represents a powerful framework for advancing drug repurposing and understanding polypharmacology. As these approaches mature, they promise to accelerate the delivery of safe, effective treatments for diverse diseases while reducing the overall costs of therapeutic development.
Chemogenomics has emerged as a pivotal discipline in modern drug discovery, systematically exploring the interaction space between small molecules and biological target families. This whitepaper examines the evolving paradigm from traditional forward and reverse chemogenomic approaches toward integrated hybrid screening strategies. By combining phenotypic and target-based screening with advanced computational methods, researchers can accelerate target identification, validation, and therapeutic development. We present quantitative comparisons of screening methodologies, detailed experimental protocols, and essential research tools that enable more efficient navigation of the chemical-biological interaction landscape. The integration of these approaches addresses critical limitations of single-method screening, particularly in complex disease contexts, offering a more comprehensive framework for identifying novel therapeutic agents and their mechanisms of action.
Chemogenomics represents a systematic framework for screening targeted chemical libraries against families of drug targets—such as GPCRs, kinases, proteases, and nuclear receptors—with the dual objectives of identifying novel drugs and elucidating new drug targets [1]. This approach leverages the fundamental principle that ligands designed for one family member often exhibit affinity for related targets, enabling parallel exploration of chemical and biological spaces [1]. The completion of the human genome project has provided an unprecedented abundance of potential therapeutic targets, making systematic approaches like chemogenomics essential for comprehensive therapeutic intervention [1].
The traditional dichotomy in chemogenomic screening distinguishes between forward chemogenomics (phenotype-based) and reverse chemogenomics (target-based) approaches [1]. Forward chemogenomics begins with a desired phenotype and identifies small molecules that induce it, subsequently determining the molecular targets responsible [1]. Conversely, reverse chemogenomics starts with a specific protein target, identifies compounds that modulate its activity, and then characterizes the resulting phenotypes in cellular or organismal models [1]. While both approaches have proven valuable, they each present distinct limitations in throughput, target identification, and physiological relevance.
This whitepaper advances the thesis that hybrid screening methodologies that integrate forward and reverse paradigms represent the future of chemogenomics. By combining the physiological relevance of phenotypic screening with the mechanistic clarity of target-based approaches, researchers can overcome the inherent limitations of either method alone. The following sections provide a comprehensive technical examination of both established and emerging hybrid screening strategies, with particular emphasis on practical implementation, quantitative comparison, and translational application in drug development.
Core Screening Paradigms: Forward vs. Reverse Chemogenomics
Forward (Classical) Chemogenomics
Forward chemogenomics employs phenotypic screening to identify compounds that induce a specific biological response without prior knowledge of the molecular target [1]. The methodological workflow begins with developing robust phenotypic assays that accurately recapitulate the disease-relevant biology, followed by screening compound libraries to identify modulators that produce the desired phenotype [1]. The primary challenge lies in designing phenotypic assays that enable direct transition from screening to target identification [1].
A key application of forward chemogenomics appears in antimicrobial discovery, where researchers developed a bivariate primary screen assessing motility and viability of filarial parasite microfilariae [79]. This approach identified 35 hit compounds from a 1,280-compound library (2.7% hit rate), with subsequent dose-response characterization revealing 13 compounds exhibiting EC50 values below 1μM for at least one phenotypic endpoint [79]. The study demonstrated that multiplexed phenotypic assessment at multiple timepoints captured non-redundant biological information, with motility and viability measurements showing high overall correlation (r = -0.84) but substantially lower correlation among hits (r = 0.33) [79].
Reverse Chemogenomics
Reverse chemogenomics begins with a validated protein target and identifies small molecules that modulate its activity in biochemical assays, then characterizes the resulting phenotypes in cellular or organismal systems [1]. This approach benefits from parallel screening capabilities and optimized lead compounds across multiple targets within the same gene family [1]. Reverse chemogenomics has been enhanced by the availability of targeted chemical libraries enriched for compounds known to interact with specific protein families [6].
In practice, reverse chemogenomics was employed to discover novel heat shock protein 90 (Hsp90) inhibitors using a yeast-based screening platform [80]. Researchers screened 3,680 compounds against Saccharomyces cerevisiae strains with differential sensitivity to Hsp90 inhibitors, using time-dependent turbidity measurements in liquid culture to quantify growth phenotypes [80]. This approach identified the known Hsp90 inhibitor macbecin and a novel chemotype (NSC145366) that subsequent biochemical characterization revealed interacts with the Hsp90 C-terminus through a mechanism distinct from classical N-terminal inhibitors [80].
Comparative Analysis of Screening Approaches
Table 1: Quantitative Comparison of Forward and Reverse Chemogenomic Screening Approaches
| Parameter | Forward Chemogenomics | Reverse Chemogenomics |
|---|---|---|
| Starting Point | Phenotype of interest [1] | Specific protein target [1] |
| Screening Context | Cellular or organismal models [79] | Biochemical or cell-based target-specific assays [80] |
| Target Identification | Required after compound identification; can be challenging [1] | Known prior to screening [1] |
| Hit Rate | 2.7% in microfilariae screen [79] | Varies by target; 0.2% in Hsp90 screen [80] |
| Physiological Relevance | High; measures integrated biological responses [79] | Variable; depends on assay design [80] |
| Throughput | Moderate; limited by complex assays [79] | High; amenable to automation [80] |
| Key Challenge | Designing assays that enable target identification [1] | Recapitulating physiological complexity [80] |
Hybrid Screening Methodologies: Integrated Approaches
Tiered Multivariate Phenotypic Screening
Advanced hybrid screening strategies employ tiered, multivariate approaches that leverage strengths of both forward and reverse paradigms. A exemplar study implemented a bivariate primary screen against filarial parasite microfilariae assessing motility and viability, followed by secondary multivariate screening against adult parasites evaluating neuromuscular function, fecundity, metabolism, and viability [79]. This approach achieved an exceptional >50% hit rate for macrofilaricidal compounds by leveraging the abundant microfilarial stage to enrich for compounds with activity against the more physiologically relevant but less accessible adult stage [79].
The methodological strength of this approach lies in its capacity to capture stage-specific and phenotype-specific compound effects, enabling identification of chemotypes with differential activity across parasite life stages. For example, the screen identified five compounds with high potency against adult parasites but low potency or slow-acting effects against microfilariae, suggesting novel mechanisms of action potentially distinct from existing anthelmintics [79]. This phenotypic precision enables more informed lead selection and prioritization for resource-intensive downstream studies.
Chemogenomic Library Design for Hybrid Screening
Effective hybrid screening requires carefully designed chemical libraries that incorporate both target coverage and chemical diversity. A recently developed chemogenomic library of 5,000 small molecules represents a diverse panel of drug targets involved in multiple biological processes and diseases, integrated within a systems pharmacology network incorporating drug-target-pathway-disease relationships [81]. This library was constructed through analysis of the ChEMBL database, KEGG pathways, Gene Ontology terms, and morphological profiling data from the Cell Painting assay [81].
For precision oncology applications, researchers developed a minimal screening library of 1,211 compounds targeting 1,386 anticancer proteins, optimized for library size, cellular activity, chemical diversity, availability, and target selectivity [33]. In a pilot screening against glioblastoma stem cells from patients, this library identified highly heterogeneous phenotypic responses across patients and molecular subtypes, demonstrating the value of targeted library design for detecting patient-specific vulnerabilities [33]. The resulting physical library of 789 compounds covered 1,320 anticancer targets while maintaining practical screening feasibility [33].
Table 2: Quantitative Performance of Hybrid Screening Platforms
| Screening Platform | Library Size | Assay Type | Hit Rate | Key Outcomes |
|---|---|---|---|---|
| Filarial Parasite Screen [79] | 1,280 compounds | Bivariate phenotypic (motility/viability) | 2.7% primary, >50% confirmed | 13 compounds with EC50 <1μM; 5 with adult-specific activity |
| Hsp90 Inhibitor Platform [80] | 3,680 compounds | Yeast growth phenotypic | 0.2% | Identified novel C-terminal Hsp90 inhibitor |
| Glioblastoma Screen [33] | 789 compounds | Image-based phenotypic (patient cells) | Patient-dependent | Identified patient-specific vulnerabilities |
| Systems Pharmacology [81] | 5,000 compounds | Multiple assay types | Network-dependent | Integrated target-phenotype-disease relationships |
Computational Integration and Target Prediction
Hybrid screening approaches increasingly incorporate computational methods to bridge chemical and biological spaces. Chemogenomics leverages deep learning approaches to model complex relationships between chemical structures and protein targets, going beyond classical QSAR methods that predict ligands for single proteins to simultaneously predict interactions across multiple targets [6]. For example, deep learning-based fragment linking methods such as SyntaLinker-Hybrid enable target-specific molecular generation through transfer learning and fragment hybridization [82].
The integration of heterogeneous data sources represents another key advancement in hybrid screening. One research platform integrated the ChEMBL database, KEGG pathways, Gene Ontology, Disease Ontology, and morphological profiling data from the Cell Painting assay within a Neo4j graph database [81]. This network pharmacology approach enables identification of proteins modulated by chemicals that correlate with specific morphological perturbations, facilitating target identification for phenotypic screening hits [81].
Experimental Protocols for Hybrid Screening
Multivariate Phenotypic Screening in Parasites
Objective: Identify compounds with macrofilaricidal activity using a tiered screening approach leveraging microfilariae for primary screening and adult parasites for secondary validation [79].
Primary Screen (Microfilariae):
- Parasite Preparation: Isolate B. malayi microfilariae from rodent hosts, followed by column filtration to remove debris and improve assay quality [79].
- Assay Setup: Dispense 100-500 mf per well in 384-well plates. Add compounds at 100μM initial concentration with appropriate controls (heat-killed mf for viability control) [79].
- Phenotypic Assessment:
- Hit Selection: Define hits as compounds producing Z-score >1 in either motility or viability measurements relative to DMSO controls [79].
Secondary Screen (Adult Parasites):
- Parasite Culture: Maintain adult B. malayi parasites in supplemented medium. Use freshly harvested parasites for all assays [79].
- Multiplexed Phenotyping:
- Motility: Quantify movement patterns using video tracking and analysis software.
- Fecundity: Assess embryo production and development through microscopic examination.
- Metabolism: Measure metabolic activity using resazurin reduction assays.
- Viability: Determine parasite survival using morphological criteria and propidium iodide exclusion [79].
- Dose-Response Characterization: Generate 8-point dose-response curves for confirmed hits. Calculate EC50 values for each phenotypic endpoint [79].
Validation: Prioritize compounds showing differential activity between life stages or distinct phenotypic profiles for further mechanistic studies and target identification.
Yeast-Based Chemogenomic Screening
Objective: Identify novel Hsp90 inhibitors using differential sensitivity of yeast deletion strains in a growth-based phenotypic screen [80].
Strain Selection and Preparation:
- Strain Collection: Select Saccharomyces cerevisiae strains with known sensitivity to Hsp90 inhibition (e.g., ydj1Δ, hsp82Δ, sst2Δ) alongside wild-type controls [80].
- Culture Conditions: Grow yeast strains in YPD medium overnight. Dilute to standardized density in minimal proline medium (MPD) containing 0.003% SDS to enhance compound permeability [80].
Screening Protocol:
- Assay Setup: Dispense 25μL diluted yeast culture into 384-well plates containing 25μL compound solution (final concentration 200μM or 40μM). Include DMSO controls and reference inhibitors (geldanamycin, radicicol) on each plate [80].
- Growth Monitoring: Seal plates with transparent tape and incubate at 30°C. Measure optical density at 600nm every hour for 48-60 hours using plate readers [80].
- Data Analysis:
- Normalize growth curves using integrals and initial optical density.
- Calculate growth rates and determine time to reach OD600 of 0.8 for each strain-compound combination.
- Compare compound sensitivity across strains to identify chemogenomic profiles [80].
Hit Confirmation:
- Dose-Response: Retest hits in dose-response format against primary screening strains and additional Hsp90-sensitive strains.
- Mechanistic Studies: Employ biochemical assays (ATPase activity, client protein maturation) and binding studies (surface plasmon resonance) to confirm direct Hsp90 interaction and characterize mechanism of inhibition [80].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Chemogenomic Screening
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Chemical Libraries | Tocriscreen 2.0 Library [79], LOPAC1280 [80], NCATS MIPE Library [81] | Provide diverse chemical matter with annotated targets for screening |
| Cell-Based Assay Systems | Haploid yeast deletion strains [80], Patient-derived glioblastoma cells [33], Filarial parasite life stages [79] | Enable phenotypic screening in disease-relevant contexts |
| Detection Reagents | ATP-based viability assays [79], Cell Painting stains [81], Resazurin metabolism assays [79] | Quantify phenotypic endpoints and cellular responses |
| Bioinformatics Tools | ChEMBL database [81], KEGG pathways [81], Neo4j graph database [81] | Integrate and analyze chemogenomic data across multiple dimensions |
| Specialized Media | Minimal proline medium (MPD) with SDS [80], Supplemented parasite culture media [79] | Optimize assay conditions for compound penetration and phenotype detection |
The integration of forward and reverse chemogenomic approaches represents a paradigm shift in drug discovery, addressing fundamental limitations of single-method screening strategies. Hybrid methodologies leverage the physiological relevance of phenotypic screening while incorporating the mechanistic insights of target-based approaches, creating a more comprehensive framework for identifying and validating novel therapeutic agents. The quantitative data and experimental protocols presented in this whitepaper demonstrate the practical implementation and substantial advantages of these integrated approaches across diverse therapeutic areas.
Future developments in chemogenomic screening will likely focus on several key areas: (1) enhanced computational prediction of target-phenotype relationships through deep learning and network pharmacology; (2) increased integration of multi-omics data to contextualize compound activity within broader biological networks; and (3) development of more sophisticated phenotypic profiling methods that capture complex disease biology. As these technologies mature, hybrid screening approaches will become increasingly central to drug discovery, enabling more efficient navigation of the complex landscape connecting chemical space to biological function and therapeutic application.
Conclusion
Forward and reverse chemogenomics are not opposing but complementary strategies that form a powerful cycle for biological discovery and therapeutic development. Forward chemogenomics excels at uncovering novel biology and unexpected drug targets by starting with a phenotypic observation, while reverse chemogenomics provides a rational, target-focused path for validating disease mechanisms and optimizing lead compounds. The convergence of these approaches with advanced computational methods, particularly AI and machine learning, is dramatically accelerating the process. Looking ahead, global open-science initiatives like Target 2035 and the development of extensive chemogenomic libraries are poised to systematically expand the druggable proteome. The future of drug discovery lies in the intelligent integration of both paradigms, leveraging large-scale, high-quality data to transform hit-finding into a predictive science and ultimately deliver novel therapeutics for diseases with unmet needs.
References
- 1. Chemogenomics [https://en.wikipedia.org/wiki/Chemogenomics]
- 2. Chemogenomics : a new approach to quick target and drug ... [https://www.linkedin.com/pulse/chemogenomics-new-approach-quick-target-drug-saveena-solanki]
- 3. The Future of Drug Discovery: Integrating Phenotypic Data ... [https://ardigen.com/the-future-of-drug-discovery-integrating-phenotypic-data-with-omics-and-ai/]
- 4. DeepDTAGen: a multitask deep learning framework for ... [https://www.nature.com/articles/s41467-025-59917-6]
- 5. Beyond Legacy Tools: Defining Modern AI Drug Discovery ... [https://www.biopharmatrend.com/business-intelligence/what-is-ai-drug-discovery/]
- 6. Chemogenomics - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/chemogenomics]
- 7. From Patterns to Pills: How Informatics Is Shaping Medicinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114992/]
- 8. Toward target 2035: EUbOPEN - a public–private ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11605244/]
- 9. A Practical Guide to Chemogenomics Data Curation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5657146/]
- 10. Advances in machine learning for optimizing ... [https://www.sciencedirect.com/science/article/pii/S1570164625000153]
- 11. A review on computer‐aided chemogenomics and drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9539342/]
- 12. Target identification and mechanism of action in chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5543995/]
- 13. Reverse Screening Methods to Search for the Protein ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2018.00138/full]
- 14. Chemogenomics: Methods and Protocols ... [https://dokumen.pub/chemogenomics-methods-and-protocols-methods-in-molecular-biology-2706-1st-ed-2023-1071633961-9781071633960.html]
- 15. Combination chemical genetics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2712875/]
- 16. Phenotypic Versus Target-Based Screening for Drug ... [https://www.technologynetworks.com/drug-discovery/articles/phenotypic-versus-target-based-screening-for-drug-discovery-300037]
- 17. Phenotypic screening [https://en.wikipedia.org/wiki/Phenotypic_screening]
- 18. Phenotypic vs. target-based drug discovery for first-in-class ... [https://pubmed.ncbi.nlm.nih.gov/23511784/]
- 19. Chemogenomics - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/chemogenomics]
- 20. Phenotypic Screening [https://www.revvity.com/category/phenotypic-screening]
- 21. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 22. Perspectives on phenotypic screening−Screen Design and ... [https://www.sciencedirect.com/science/article/pii/S247255522400008X]
- 23. Global Approach to Biomedicine: Functional Genomics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6008969/]
- 24. A Comprehensive Review of Bioinformatics Tools for Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11353282/]
- 25. : an emerging strategy for... | Nature Reviews Genetics Chemogenomics [https://www.nature.com/articles/nrg1317?error=cookies_not_supported]
- 26. Development of a chemogenomics library for phenotypic ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00569-1]
- 27. TARGET VALIDATION IN CHEMOGENOMICS [https://www.sciencedirect.com/science/article/abs/pii/B9780123693938500031]
- 28. Chemogenomic Approaches for Revealing Drug Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8844939/]
- 29. ExCAPE-DB: an integrated large scale dataset facilitating Big ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0203-5]
- 30. Chemical genetics in drug discovery [https://www.sciencedirect.com/science/article/pii/S2452310017301233]
- 31. Composition and applications of focus libraries to phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4109565/]
- 32. Quantitative high-throughput screening data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375054/]
- 33. Chemogenomic library design strategies for precision ... [https://www.sciencedirect.com/science/article/pii/S2589004223012865]
- 34. Systems Pharmacology for Investigation of the Mechanisms of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6657703/]
- 35. Chemogenomics: Is A Promising Area for Drug Target and ... [https://article.innovationforever.com/JMBDD/20220173.html]
- 36. Chemogenomics approaches to rationalising compound ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3606204/]
- 37. G protein-coupled receptors (GPCRs): advances in ... [https://www.nature.com/articles/s41392-024-01803-6]
- 38. Discovery of GPCR ligands for probing signal transduction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4246677/]
- 39. Emerging paradigms for target discovery of traditional medicines [https://pmc.ncbi.nlm.nih.gov/articles/PMC11910885/]
- 40. Intracellular GPCR modulators enable precision ... [https://www.nature.com/articles/s44386-025-00011-8]
- 41. Visualization of Activity Landscapes and Chemogenomics ... [https://pubmed.ncbi.nlm.nih.gov/27481141/]
- 42. The Deconvolution Revolution: Strategies for Target ... [https://momentum.bio/content/the-deconvolution-revolution-strategies-for-target-identification/]
- 43. Combining experimental strategies for successful target ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644620303731]
- 44. Pattern Recognition Algorithms in Pharmacogenomics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12655502/]
- 45. The limitations of small molecule and genetic screening in ... [https://www.sciencedirect.com/science/article/pii/S2451945625003459?dgcid=rss_sd_all]
- 46. Chemogenomics for steroid hormone receptors (NR3) [https://www.nature.com/articles/s42004-025-01427-z]
- 47. Chemogenomics for drug discovery: clinical molecules from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8341094/]
- 48. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 49. Convergence of Computer-Aided Drug Discovery and ... [https://www.sciencedirect.com/science/article/pii/S2773216925000388]
- 50. Machine Learning in Drug Discovery: A Review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8356896/]
- 51. Machine learning in preclinical drug discovery [https://www.nature.com/articles/s41589-024-01679-1]
- 52. DTIAM: a unified framework for predicting drug-target ... [https://www.nature.com/articles/s41467-025-57828-0]
- 53. Artificial Intelligence (AI) Applications in Drug Discovery and Drug Delivery: Revolutionizing Personalized Medicine - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11510778/]
- 54. Application of Artificial Intelligence In Drug-target Interactions Prediction: A Review | npj Biomedical Innovations [https://www.nature.com/articles/s44385-024-00003-9]
- 55. Why Cheminformatics is Important for Organic Chemists in ... [https://neovarsity.org/blogs/cheminformatics-for-organic-chemists]
- 56. Time For a New Adventure - Practical Cheminformatics [https://patwalters.github.io/Time-For-a-New-Adventure/]
- 57. EUbOPEN: Homepage [https://www.eubopen.org/]
- 58. PubChem 2025 update - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701573/?utm_source=ncbi_twitter&utm_medium=referral&utm_campaign=pmc-nar-20250315]
- 59. How Cheminformatics is Used in Drug Discovery in 2025? [https://neovarsity.org/blogs/how-cheminformatics-is-used-in-drug-discovery]
- 60. A Curated List of Cheminformatics Software and Libraries [https://neovarsity.org/blogs/curated-list-cheminformatics-software-libraries]
- 61. the emerging impact of chemoinformatics in chemistry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333164/]
- 62. Recommendations for bioinformatics in clinical practice [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-025-01543-4]
- 63. Unlocking the Potential of Bioinformatics and ... [https://www.desertsci.com/2025/05/19/unlocking-the-potential-of-bioinformatics-and-cheminformatics-in-structure-based-drug-design/]
- 64. Bioinformatics in 2025: Applications & Insights [https://riveraxe.com/what-is-bioinformatics-and-its-application/]
- 65. The 2025 Bioinformatics Toolkit: From Sample Prep to AI- ... [https://blog.omni-inc.com/blog/the-2025-bioinformatics-toolkit-from-sample-prep-to-ai-driven-analysis]
- 66. Comparative Profiling and Chemogenomics Application of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12516683/]
- 67. Toward target 2035: EUbOPEN - a public–private partnership ... [https://pubs.rsc.org/en/content/articlehtml/2025/md/d4md00735b]
- 68. How new approach methodologies are reshaping drug ... [https://www.mckinsey.com/industries/life-sciences/our-insights/the-synthesis/how-new-approach-methodologies-are-reshaping-drug-discovery]
- 69. Machine Learning for Multi-Target Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473769/]
- 70. Chemical genetics in drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7371212/]
- 71. Phenotypic Drug Discovery: Recent successes, lessons ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9708951/]
- 72. Chemogenomic approaches to rational drug design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1978269/]
- 73. Review Hit Triage and Validation in Phenotypic Screening [https://www.sciencedirect.com/science/article/pii/S2451945620303317]
- 74. AΙ-Driven Drug Repurposing: Applications and Challenges [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641654/]
- 75. Literature data-based de novo candidates for drug repurposing [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06237-7]
- 76. Data-driven strategies for drug repurposing - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641610/]
- 77. A standard database for drug repositioning | Scientific Data [https://www.nature.com/articles/sdata201729]
- 78. Therapeutic innovation in drug repurposing: Challenges ... [https://www.sciencedirect.com/science/article/pii/S1359644625001035]
- 79. Multivariate chemogenomic screening prioritizes new ... [https://www.nature.com/articles/s42003-023-04435-8]
- 80. A Chemogenomic Screening Platform Used to Identify ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6521697/]
- 81. Development of a chemogenomics library for phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8611952/]
- 82. SyntaLinker-Hybrid: A deep learning approach for target ... [https://www.sciencedirect.com/science/article/pii/S266731852200006X]