Data Harmonization in Multi-Omics Studies: 2025 Best Practices for Robust Integration and Clinical Translation
This article provides a comprehensive guide to data harmonization best practices tailored for researchers, scientists, and drug development professionals working with multi-omics data.
Data Harmonization in Multi-Omics Studies: 2025 Best Practices for Robust Integration and Clinical Translation
Abstract
This article provides a comprehensive guide to data harmonization best practices tailored for researchers, scientists, and drug development professionals working with multi-omics data. It covers the foundational principles of multi-omics integration, explores advanced methodological strategies for combining diverse datasets, offers solutions for common troubleshooting and optimization challenges, and outlines rigorous validation and comparative analysis frameworks. By addressing these four core intents, the article aims to equip practitioners with the knowledge to transform complex, heterogeneous biological data into reliable, actionable insights for precision medicine and therapeutic discovery.
Laying the Groundwork: Core Principles and the Imperative for Multi-Omics Harmonization
Defining Data Harmonization in the Multi-Omics Context
Frequently Asked Questions (FAQs)
1. What is the fundamental difference between data harmonization and data integration in multi-omics studies?
Data harmonization is the crucial preparatory step that ensures different omics datasets are comparable and ready for integration. It involves mapping data to common ontologies, normalizing data to comparable scales or units, and applying consistent filtering criteria to mitigate technical variations like batch effects [1]. Data integration, conversely, is the subsequent step of jointly analyzing these harmonized datasets using statistical or machine learning methods (e.g., MOFA, DIABLO) to extract biological insights [2]. Simply put, harmonization makes the data uniform, while integration finds the meaning in the combined data.
2. How can I check if my datasets are compatible for multi-omics integration?
Before integration, verify the following aspects of your experimental design [1]:
- Sample Context: Ensure datasets originate from the same biological sample type (e.g., disease tissue vs. healthy control, same cell population).
- Population Consistency: Confirm that samples are from a comparable population regarding factors like gender, age, or treatment history.
- Metadata Alignment: Carefully read the metadata for each dataset to ensure key variables (e.g., clinical outcomes, experimental conditions) are defined and measured consistently across studies.
3. What are the best practices for handling missing data in multi-omics datasets?
Missing data is a common challenge, often arising from technological limits where molecules like proteins might be undetectable in one sample but present in another [2]. Best practices include:
- Generative Models: Advanced AI methods, such as Variational Autoencoders (VAEs) or Generative Adversarial Networks (GANs), can learn the underlying data distribution to impute plausible values for missing data points [3].
- Factorization Methods: Tools like MOFA (Multi-Omics Factor Analysis) are designed to handle missing values by inferring latent factors that explain the observed data, without requiring complete datasets [2].
- Quality Filtering: As a foundational step, prioritize data from carefully quality-controlled (QC-ed) studies to minimize non-random missingness from poor sample quality [1].
4. Which integration method should I choose for my specific biological question?
The choice of integration method is not one-size-fits-all and should be guided by your research goal. The table below summarizes the purpose of several state-of-the-art methods.
| Method | Primary Purpose | Key Characteristics |
|---|---|---|
| MOFA [2] | Unsupervised discovery of latent factors driving variation across omics layers. | Probabilistic, Bayesian framework; identifies shared and data-specific factors; does not require a pre-defined outcome. |
| DIABLO [2] | Supervised integration for biomarker discovery and phenotype prediction. | Uses known phenotype labels; performs feature selection to identify molecules predictive of a specific category (e.g., disease vs. healthy). |
| SNF [2] [4] | Unsupervised sample clustering and network-based fusion. | Constructs and fuses sample-similarity networks from each omics data type to identify patient subgroups. |
| Correlation Networks [4] | Uncover relationships between different molecular entities (e.g., genes and metabolites). | Uses statistical correlations (e.g., Pearson) to build interaction networks, helping identify key regulatory nodes and pathways. |
5. How can I address the "batch effect" problem when combining datasets from different studies or labs?
Batch effects, where technical variations obscure biological signals, are a major harmonization hurdle. Key strategies include:
- Standardization and Transformation: Apply consistent normalization methods across all datasets. Transforming data to a ranking system is a common practice to alleviate batch effects [1].
- Similarity Network Fusion (SNF): This method can be effective as it fuses data based on sample-similarity patterns, which can be more robust to batch effects than raw data integration [2] [4].
- Data Transformation: Normalize data to a consistent scale (e.g., 0-1) before integration to make them comparable, a technique often used in target prioritization pipelines [1].
Troubleshooting Guides
Issue: Incompatible Data Formats and Ontologies
Problem: You have collected transcriptomics and metabolomics data, but they are in different formats (e.g., raw count matrices vs. peak intensity tables), use different gene/protein identifiers, and lack standardized metadata.
Solution: Implement a comprehensive standardization and harmonization workflow.
Methodology:
- Format Conversion: Convert all data into a matrix format where rows are features (e.g., genes, proteins) and columns are samples.
- Identifier Mapping: Map all gene, protein, and metabolite identifiers to a consistent ontology or database (e.g., Ensembl IDs for genes, HMDB IDs for metabolites).
- Metadata Annotation: Create a unified metadata table for all samples, ensuring clinical or phenotypic terms are drawn from controlled vocabularies.
- Normalization: Apply appropriate normalization techniques for each data type (e.g., TPM for RNA-seq, quantile normalization for proteomics) to make distributions comparable.
Issue: High-Dimensionality and Data Sparsity
Problem: Your integrated dataset has thousands of molecular features (high dimensionality) but only a limited number of biological samples, and some data types (e.g., metabolomics) are inherently sparse, leading to overfitting and poor model performance.
Solution: Employ dimensionality reduction and feature selection techniques.
Methodology:
- Feature Filtering: Remove low-variance features and those with a high proportion of missing values.
- Factorization / Latent Variable Models: Use methods like MOFA to reduce dimensionality by inferring a small number of latent factors that capture the major sources of biological variation across all omics datasets [2] [3].
- Supervised Feature Selection: When a phenotype is known, use supervised methods like DIABLO, which incorporates penalization (e.g., Lasso) to select only the most informative features for integration and prediction [2].
- AI-Driven Integration: Leverage deep learning architectures like autoencoders to learn compressed, lower-dimensional representations of the data that are suitable for downstream tasks [3].
Issue: Interpreting Biologically Meaningful Results from Integrated Models
Problem: After running an integration model, you have a list of features or factors but struggle to translate these statistical outputs into actionable biological hypotheses.
Solution: Combine integration outputs with downstream functional analysis.
Methodology:
- Factor Interpretation (for MOFA): Examine the top features (genes, proteins) with the highest weights ("loadings") for each inferred factor. Then, perform pathway enrichment analysis on these top-feature sets.
- Network Integration: Map the results onto shared biochemical networks. For example, connect a prioritized transcription factor (from transcriptomics) to the transcripts it regulates and the associated metabolites from the metabolic pathways it influences [4] [5].
- Multi-Omics Pathway Analysis: Use pathway enrichment methods that are specifically designed for and can incorporate multiple types of omics data simultaneously, rather than analyzing each result in isolation [4] [1].
The Scientist's Toolkit: Essential Research Reagents & Solutions
The following table details key computational tools and resources essential for conducting robust multi-omics data harmonization and integration.
| Tool/Resource Name | Function | Application in Harmonization/Integration |
|---|---|---|
| MOFA+ [2] | Unsupervised multi-omics data integration | Discovers latent factors that capture the main sources of variation across multiple omics datasets. Ideal for exploratory analysis. |
| DIABLO [2] | Supervised multi-omics integration | Integrates data in relation to a categorical outcome for biomarker discovery and sample classification. |
| WGCNA [4] | Weighted Gene Co-expression Network Analysis | Identifies modules of highly correlated features; modules can be related to external traits or other omics data. |
| Cytoscape [4] | Network visualization and analysis | Visualizes complex interaction networks (e.g., gene-metabolite networks) derived from integrated data. |
| TCGA [2] [3] | Publicly available multi-omics database | Provides a vast resource of matched multi-omics data for method development, validation, and benchmarking. |
| Omics Playground [2] | Integrated analysis platform | Offers a code-free interface with multiple state-of-the-art integration methods and visualization capabilities. |
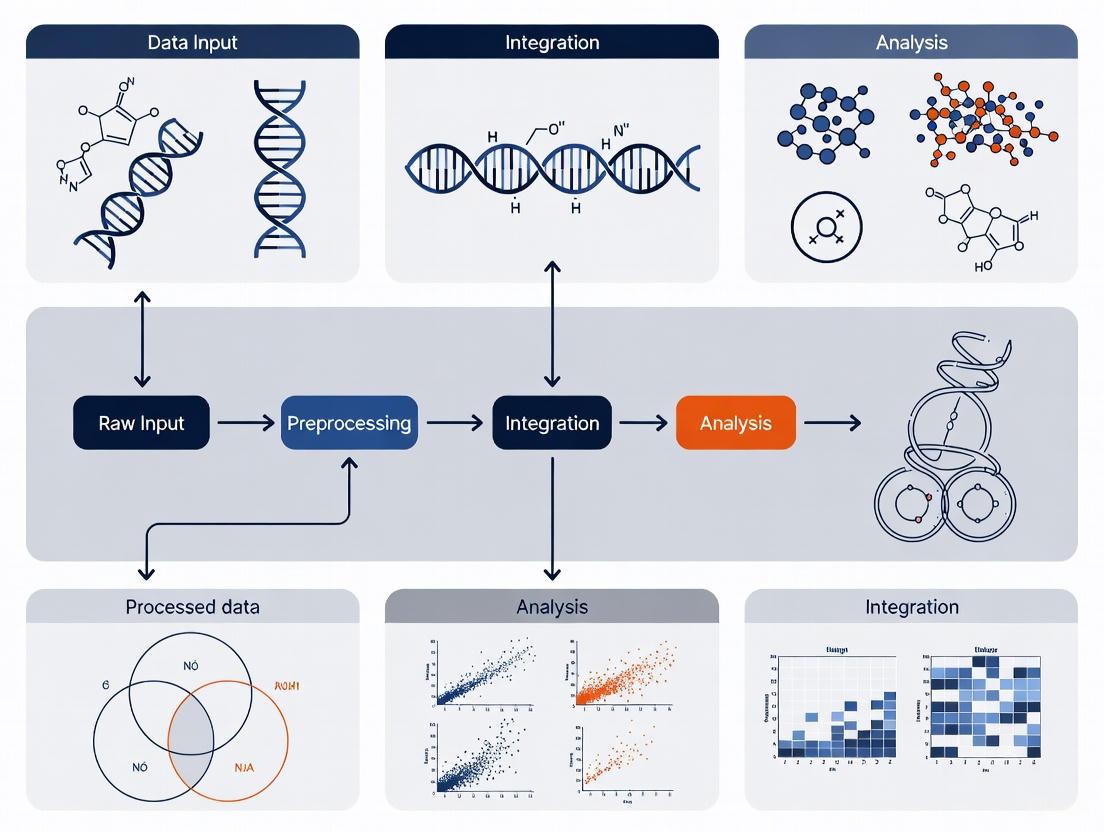
Conceptual Framework & Data Harmonization Strategies
What is multi-omics data integration and why is harmonization critical?
Multi-omics data integration involves combining and collectively analyzing disparate biological data layers, such as genomics, transcriptomics, proteomics, and metabolomics, to gain a comprehensive understanding of complex biological systems [6]. Data harmonization is the process of reconciling these various types, levels, and sources of data into formats that are compatible and comparable, making them useful for integrated analysis and decision-making [7]. This is essential because without effective harmonization, multi-omics analysis becomes more complex and resource-intensive without proportional gains in insight or productivity [8].
What are the primary strategies for integrating multi-omics data?
The integration of vertical or heterogeneous data (data from different omics levels) can be approached through several distinct strategies [8]. The choice of strategy depends on the biological question, data characteristics, and computational resources.
Table 1: Overview of Multi-Omics Data Integration Strategies
| Integration Strategy | Description | Key Advantages | Key Limitations |
|---|---|---|---|
| Early Integration | Concatenates all omics datasets into a single matrix prior to analysis [8]. | Simple and easy to implement [8]. | Creates a complex, high-dimensional matrix that is noisy and discounts data distribution differences [8]. |
| Mixed Integration | Separately transforms each dataset into a new representation before combining them [8]. | Reduces noise, dimensionality, and dataset heterogeneities [8]. | - |
| Intermediate Integration | Simultaneously integrates datasets to output common and omics-specific representations [8]. | Captures interactions between omics layers [8]. | Often requires robust pre-processing to handle data heterogeneity [8]. |
| Late Integration | Analyzes each omics dataset separately and combines the final predictions or results [8]. | Circumvents challenges of assembling different datatypes [8]. | Does not capture inter-omics interactions during the analysis [8]. |
| Hierarchical Integration | Focuses on including prior knowledge of regulatory relationships between omics layers [8]. | Truly embodies the intent of trans-omics analysis [8]. | A nascent field; methods are often less generalizable [8]. |
The following diagram illustrates the logical flow and differences between these primary integration strategies:
Troubleshooting Common Multi-Omics Challenges
How do I handle missing values and the High Dimension Low Sample Size (HDLSS) problem?
Problem: Omics datasets often contain missing values due to technical limitations, and frequently have thousands of variables (e.g., genes, proteins) but only a small number of samples [8]. This HDLSS problem can cause machine learning algorithms to overfit, reducing their generalizability [8].
Solutions:
- Missing Data: Implement an additional imputation process to infer missing values in incomplete datasets before applying statistical analyses [8]. The choice of imputation method (e.g., mean, k-nearest neighbors, model-based) should be carefully considered based on the nature of the missingness.
- HDLSS & Overfitting: Employ dimensionality reduction techniques (e.g., PCA, autoencoders) or feature selection methods to reduce the number of variables. Use regularization techniques (e.g., Lasso, Ridge regression) within your models and always validate models using held-out test sets or cross-validation to ensure generalizability [8].
Our data is heterogeneous and lacks pre-processing standards. How can we harmonize it effectively?
Problem: The sheer heterogeneity of omics data—comprising different data modalities, distributions, and types—poses a significant challenge. The absence of standardized pre-processing protocols means each data type requires tailored processing, introducing variability [8] [2].
Solutions:
- Adopt Established Data Standards: Utilize existing minimum information standards and data formats developed by the omics communities. Examples include:
- Flexible Harmonization: Recognize that stringent harmonization (using identical measures) is not always possible. Instead, aim for flexible harmonization, which ensures datasets are inferentially equivalent even if not identical, and transform them into a common format [7]. This involves resolving heterogeneity across three dimensions:
- Syntax: Convert data into a common technical format (e.g.,
.csv,.json). - Structure: Reconcile how variables relate to each other (e.g., from event data to panel data).
- Semantics: Carefully map the intended meaning of variables and ensure consistent operationalization of concepts across datasets [7].
- Syntax: Convert data into a common technical format (e.g.,
- Common Data Elements (CDEs): For clinical and cohort data, develop and use CDEs—standardized concepts that precisely define a question with a specified set of responses—to promote standardized data capture and retrospective harmonization [10].
How do we choose the right integration method from the many available?
Problem: A wide array of computational tools exists for multi-omics integration, leading to confusion about which method is best suited for a specific dataset or biological objective [11] [2].
Solutions:
- Align Method with Objective: The choice of integration tool should be driven by the primary scientific objective of your study [11]. The table below maps common objectives to suitable tools and methods.
Table 2: Matching Integration Tools to Scientific Objectives
| Scientific Objective | Recommended Method Type | Example Tools & Brief Description |
|---|---|---|
| Subtype Identification | Unsupervised methods that group samples based on shared multi-omics profiles [11]. | MOFA+ [2]: Unsupervised factor analysis to uncover latent sources of variation. SNF [2]: Fuses sample-similarity networks from each omics layer. |
| Detect Disease-Associated Molecular Patterns | Supervised or unsupervised methods that identify features correlated with a phenotype [11]. | DIABLO [2]: Supervised method for biomarker discovery and classification. MCIA [2]: Multivariate method to find correlated patterns across omics. |
| Understand Regulatory Processes | Methods that can model interactions and hierarchies between omics layers [11]. | Hierarchical Integration [8]: Incorporates prior knowledge of regulatory relationships (e.g., genomic variants influencing transcript levels). |
| Diagnosis/Prognosis & Drug Response Prediction | Supervised methods that build predictive models from multi-omics input [11]. | DIABLO [2]: Can be used for classification. Various machine learning models (e.g., random forests, neural networks) using late or intermediate integration. |
- Use Multiple Methods: For robust findings, consider using multiple integration methods to see if they yield consistent results [2].
- Leverage Validated Platforms: To reduce the bioinformatics bottleneck, consider using integrated analysis platforms like Omics Playground, which provide access to multiple state-of-the-art methods through a user-friendly interface [2].
How can we ensure the quality and biological relevance of our integrated results?
Problem: The outputs of integration algorithms can be statistically complex and challenging to interpret, with a risk of drawing spurious biological conclusions [2].
Solutions:
- Robust Validation: Implement a rigorous validation workflow. For subtype identification, validate clusters by assessing survival differences, clinical enrichment, or using external datasets. For supervised models, use held-out test sets and cross-validation [11].
- Downstream Biological Analysis: Use pathway and network analysis tools on the features (e.g., genes, proteins) highlighted by the integration model to place them in a functional biological context [2].
- Iterative Harmonization Checks: During data preparation, programmatically validate harmonized data. Check for adherence to controlled response options, data structure and format, value ranges, and conditional field consistency. Assign "Pass," "Fail," or "Warning" statuses to fields for review [10].
The following workflow outlines a robust process for preparing and validating harmonized data:
Experimental Protocols & Methodologies
Protocol for a Retrospective Multi-Omics Data Harmonization Project
This protocol is adapted from large-scale consortia experiences, such as the NHLBI CONNECTS program [10].
Objective: To harmonize pre-existing multi-omics and clinical datasets from different studies or cohorts into a FAIR (Findable, Accessible, Interoperable, Reusable) resource for integrated analysis.
Materials:
- Input Data: Raw multi-omics data files (e.g., FASTQ, BAM, abundance matrices) and associated clinical/ phenotypic data from multiple sources.
- Computing Infrastructure: High-performance computing or cloud-based environment (e.g., NHLBI BioData Catalyst) with sufficient storage and processing power.
- Software/Tools: Statistical programming environments (e.g., R, Python, SAS), data validation scripts, and potentially a metadata management tool.
Step-by-Step Methodology:
- Project Scoping & Team Formation:
- Define the research objectives and the specific omics datasets to be included.
- Assemble a multidisciplinary harmonization team including data managers, biostatisticians, bioinformaticians, and domain scientists [10].
Develop a Harmonization Data Dictionary:
- Define the target Common Data Elements (CDEs) that all data will be mapped to. This includes precisely defining each variable and its allowed values [10].
- Create a harmonization template that guides mappers on how to transform original study variables to the CDEs.
Execute Variable Mapping and Transformation:
- Data managers and statisticians from each study team map their native variables to the target CDEs. This process requires careful consideration of content equivalence [10].
- Programmatically transform the raw study data according to the mapping instructions. This is often done using scripts in R, Python, or SAS [10].
Automated and Manual Validation:
- Run automated validation scripts (e.g., in R) to assess the harmonized data [10]. Checks should include:
- Data structure and format (type, length).
- Adherence to controlled terminologies.
- Plausibility of value ranges and handling of missing data.
- Conditional logic (e.g., if variable A is present, variable B must also be present).
- Assign "Pass," "Fail," or "Warning" status to each field. Manually review and resolve all failures and warnings [10].
- Run automated validation scripts (e.g., in R) to assess the harmonized data [10]. Checks should include:
Data Packaging and Sharing:
- Export the validated, harmonized data into widely accessible formats (e.g., comma-delimited files).
- Prepare comprehensive metadata and documentation describing the harmonization process, assumptions, and limitations.
- Deposit both the raw and harmonized datasets, along with documentation, into a designated repository or cloud ecosystem (e.g., BioData Catalyst) to create a FAIR resource [10].
Visualization & Workflow Diagrams
Multi-Omics FAIR Data Generation Workflow
This diagram visualizes the end-to-end process of generating a standardized, harmonized multi-omics dataset ready for integration and analysis.
The Scientist's Toolkit: Research Reagent Solutions
Public Data Repositories & Knowledgebases
Table 3: Key Public Resources for Multi-Omics Research
| Resource Name | Type | Omics Content | Link |
|---|---|---|---|
| The Cancer Genome Atlas (TCGA) | Repository | Genomics, epigenomics, transcriptomics, proteomics [11] | portal.gdc.cancer.gov |
| Answer ALS | Repository | Whole-genome sequencing, RNA transcriptomics, ATAC-sequencing, proteomics, deep clinical data [11] | dataportal.answerals.org |
| jMorp | Database/ Repository | Genomics, methylomics, transcriptomics, metabolomics [11] | jmorp.megabank.tohoku.ac.jp |
| Fibromine | Database | Transcriptomics and proteomics data focused on fibrosis [11] | fibromine.com |
Computational Tools & Software
Table 4: Essential Tools for Multi-Omics Data Integration
| Tool Name | Category | Primary Function | Key Features |
|---|---|---|---|
| MOFA+ | Integration Tool | Unsupervised discovery of latent factors across multi-omics data [2]. | Probabilistic Bayesian framework; identifies shared and specific sources of variation [2]. |
| DIABLO | Integration Tool | Supervised integration for biomarker discovery and classification [2]. | Uses multiblock sPLS-DA; integrates data in relation to a categorical outcome [2]. |
| SNF | Integration Tool | Fuses sample-similarity networks from different omics types [2]. | Network-based; captures shared cross-sample similarity patterns [2]. |
| OmicsIntegrator | Utility Tool | Streamlines the process of harmonizing and integrating multi-omics datasets [6]. | Robust data integration capabilities [6]. |
| OmicsPlayground | Analysis Platform | Provides an all-in-one, code-free interface for multi-omics analysis [2]. | Integrates multiple state-of-the-art methods (MOFA, DIABLO, SNF) with visualization [2]. |
Troubleshooting Guide: Common Multi-Omics Data Harmonization Issues
This guide addresses frequent challenges encountered during multi-omics experiments, providing step-by-step solutions to ensure robust and reproducible data integration.
FAQ 1: My multi-omics datasets are in different formats and scales. How do I make them compatible for integration?
- Problem: Data from genomics, transcriptomics, and proteomics platforms arrive in disparate formats (e.g., FASTQ, BAM, raw mass spectrometry counts) with different measurement units and scales, making direct integration impossible [12] [2].
- Diagnosis: This is a standard pre-processing issue requiring data harmonization. Confirm the issue by checking for varying data distributions and value ranges across your datasets.
- Solution: Implement a standardized data harmonization pipeline [13] [14].
- Step 1: Data Acquisition & Extraction: Identify and collect all relevant data sources, including databases, APIs, and spreadsheets, noting their original formats [14].
- Step 2: Mapping: Create a unified data model or schema that defines common data elements, types, and relationships all data must follow [13] [14].
- Step 3: Ingest and Clean: Ingest raw data and clean it by removing errors, redundancies, and missing values. Normalize units, date formats, and naming conventions [13] [14].
- Step 4: Harmonize and Evaluate: Apply the defined schema to transform the raw data. This includes critical steps like normalization (e.g., using TPM for RNA-seq, FPKM for transcriptomics) to account for technical variations and batch effect correction (e.g., with tools like ComBat) to remove non-biological noise introduced by different technicians, reagents, or processing times [15] [12].
- Step 5: Deployment: Store the harmonized data in a centralized repository like a data warehouse or lake, making it accessible for analysis [13].
FAQ 2: After integration, my results are dominated by technical noise, not biological signals. What went wrong?
- Problem: The final integrated dataset or model is skewed by "batch effects" or other technical artifacts, leading to spurious conclusions [12] [2].
- Diagnosis: This indicates inadequate correction for batch effects during the pre-processing/harmonization phase. Diagnose by using Principal Component Analysis (PCA) to see if samples cluster more by processing batch than by biological group.
- Solution:
- Proactive Design: During experimental design, randomize samples across processing batches whenever possible [12].
- Statistical Correction: Apply batch effect correction algorithms after normalization but before data integration. Common methods include ComBat, Harmony, or ARSyN [12].
- Validation: Always validate that the correction worked by repeating the PCA to confirm that biological groups are now the primary source of variation.
FAQ 3: I have missing data for some omics layers in a subset of my samples. Can I still perform an integrated analysis?
- Problem: The dataset is incomplete, with some samples lacking data for one or more omics modalities (e.g., a patient has genomic data but is missing proteomic measurements) [12].
- Diagnosis: This is a common scenario in multi-omics studies, especially with clinical samples. Using only complete cases can severely bias your analysis and reduce statistical power.
- Solution: Choose an integration strategy and tools that are robust to missing data.
- Use "Late Integration" Methods: These methods build separate models for each complete omics dataset and then combine the predictions, making them naturally handle missingness [12].
- Employ Robust Imputation: Use imputation methods to estimate missing values. For example, k-nearest neighbors (k-NN) imputation can estimate a missing proteomic profile based on the profiles of samples with similar genomic and transcriptomic data [12].
- Leverage Specific Algorithms: Some multi-omics algorithms, like Multi-Omics Factor Analysis (MOFA), are designed to handle missing data by learning a latent representation from the available measurements [2].
FAQ 4: How do I choose the right data integration method for my specific biological question?
- Problem: With many multi-omics integration methods available (e.g., MOFA, DIABLO, SNF), selecting the most appropriate one is confusing [2].
- Diagnosis: The optimal method depends on your study's goal, data structure (matched vs. unmatched samples), and whether you have a specific outcome variable to predict [12] [2].
- Solution: Select your method based on the experimental goal, as summarized in the table below.
| Integration Method | Best For This Goal | Key Principle | Advantages |
|---|---|---|---|
| MOFA [2] | Unsupervised exploration; identifying latent factors that drive variation across omics layers. | Uses a Bayesian framework to infer sources of variation (factors) shared across multiple omics datasets. | Unsupervised; does not require sample labels. Handles missing data well. |
| DIABLO [2] | Supervised biomarker discovery; classifying patient groups (e.g., disease vs. healthy). | Uses a supervised, multi-block classification method to identify features that discriminate between predefined groups. | Ideal for prediction and biomarker identification. |
| SNF [12] [2] | Disease subtyping; integrating data from different sample sets. | Constructs and fuses sample-similarity networks from each omics data type into a single network. | Effective for identifying disease subtypes. Works well with unmatched data. |
FAQ 5: The results from my integrated analysis are difficult to interpret biologically. How can I translate them into insights?
- Problem: The output of a complex integration model (especially AI/ML models) is a "black box," providing patterns or feature lists without clear biological meaning [16].
- Diagnosis: This is a key bottleneck in multi-omics. The solution lies in post-integration biological interpretation.
- Solution:
- Pathway & Enrichment Analysis: Input the list of key features (genes, proteins, metabolites) identified by your model into enrichment tools (e.g., g:Profiler, Enrichr) to see if they cluster in known biological pathways [2].
- Network Integration: Map your results onto shared biochemical networks. Connect analytes (e.g., genes, proteins, metabolites) based on known interactions (e.g., a transcription factor to the transcript it regulates) to improve mechanistic understanding [5].
- Use Interpretable Models: Prioritize models that provide interpretable outputs. For instance, MOFA reveals which factors are important and which omics layers they affect, while DIABLO shows which features are most discriminative for a class [2] [16].
The Scientist's Toolkit: Essential Reagents & Materials for Multi-Omics
The following table details key reagents and solutions critical for generating robust multi-omics data, the quality of which directly impacts downstream harmonization success [15].
| Research Reagent / Material | Function in Multi-Omics Workflow |
|---|---|
| Next-Generation Sequencing (NGS) Library Prep Kits | Prepares DNA or RNA samples for sequencing by fragmenting, amplifying, and adding platform-specific adapters. Essential for genomics, epigenomics, and transcriptomics data generation. |
| Mass Spectrometry Grade Solvents & Enzymes | High-purity solvents (e.g., acetonitrile, methanol) and enzymes (e.g., trypsin) are critical for reproducible proteomics and metabolomics sample preparation and analysis, minimizing background noise. |
| Single-Cell Barcoding Reagents | Unique molecular identifiers (UMIs) and cell barcodes are used in single-cell RNA-seq (e.g., 10x Genomics) to tag molecules from individual cells, allowing for sample multiplexing and accurate transcript counting. |
| Antibodies for Protein Assays | Used in proteomics techniques like Western blot, immunoassay, or multiplexed panels (Olink, SomaScan) to specifically target and quantify protein abundance and post-translational modifications. |
| Bisulfite Conversion Reagent | Chemically modifies unmethylated cytosines in DNA to uracils, allowing for subsequent sequencing to determine genome-wide methylation patterns in epigenomics studies. |
| Cross-Linking Reagents | Chemicals like formaldehyde are used in techniques such as ChIP-seq (Chromatin Immunoprecipitation) to freeze protein-DNA interactions, enabling the study of the epigenome and transcriptome regulation. |
Experimental Protocol: A Standardized Multi-Omics Data Harmonization Workflow
This protocol outlines a generalized methodology for harmonizing disparate omics datasets, such as those from transcriptomics and proteomics, into a unified analysis-ready format [15] [13] [14].
1. Objective: To standardize, clean, and integrate raw data from multiple omics platforms into a cohesive dataset for downstream integrated analysis (e.g., using MOFA, DIABLO, or ML models).
2. Materials & Software:
- Input Data: Raw or pre-processed data matrices from various omics platforms (e.g., RNA-seq count matrix, proteomics intensity data).
- Computing Environment: R, Python, or a specialized platform like Omics Playground [2].
- Key R/Python Packages:
limma(ComBat),sva,mixOmics,MOFA2,INTEGRATE[15] [2].
3. Procedure:
- Step 1: Data Acquisition and Profiling
- Step 2: Schema Definition and Mapping
- Define a unified target schema. This includes deciding on common sample identifiers, feature naming conventions (e.g., using standard gene symbols), and data formats [13].
- Map the fields from each source dataset to the target schema.
- Step 3: Data Cleaning and Normalization
- Clean: Remove duplicates, handle missing values (e.g., via imputation or filtering), and correct obvious errors [14].
- Normalize: Apply platform-specific normalization to make data comparable within each omics type. For example:
- Step 4: Batch Effect Correction and Harmonization
- Step 5: Validation and Deployment
- Evaluate: Use visualization (e.g., PCA plots) to confirm that technical batch effects are minimized and biological signals are preserved.
- Deploy: Output the final harmonized data in an agreed-upon format (e.g., an H5 file, or multiple CSV files with aligned samples) and store it in a centralized system for analysis [13] [14].
4. Diagram: Multi-Omics Harmonization Workflow The following diagram visualizes the core steps of the data harmonization protocol.
Multi-Omics Integration Strategies at a Glance
The timing of data integration is a critical strategic decision. The table below compares the three primary approaches, which are also visualized in the subsequent diagram [12].
| Strategy | Timing | Advantages | Disadvantages |
|---|---|---|---|
| Early Integration | Data is merged before analysis. | Captures all possible cross-omics interactions; preserves raw information. | Extremely high dimensionality; computationally intensive; prone to noise. |
| Intermediate Integration | Data is transformed, then merged during analysis. | Reduces complexity; can incorporate biological context (e.g., networks). | May lose some raw information; requires careful method selection. |
| Late Integration | Models are built on each data type and merged after analysis. | Handles missing data well; computationally efficient; robust. | May miss subtle cross-omics interactions captured only by joint analysis. |
Diagram: Multi-Omics Integration Strategies
Adopting the FAIR Principles for Findable, Accessible, Interoperable, and Reusable Data
FAQs: Core FAIR Principles in Multi-Omics
What are the FAIR Data Principles and why are they critical for multi-omics research?
The FAIR Guiding Principles are a set of guidelines established in 2016 to improve the Findability, Accessibility, Interoperability, and Reuse of digital assets and data [17] [18]. In multi-omics studies, which involve integrating massive, complex datasets from genomics, transcriptomics, proteomics, and metabolomics, adhering to these principles is not merely beneficial—it is essential. FAIR provides the framework to manage the volume, velocity, and variety of multi-omics data, ensuring it can be discovered, integrated, and repurposed by both humans and computational systems to accelerate scientific discovery [5] [12] [19].
How is 'Interoperability' specifically achieved for heterogeneous omics data?
Achieving interoperability requires a multi-faceted approach centered on standardization. This involves:
- Standardized Vocabularies and Ontologies: Using shared, machine-readable languages and controlled vocabularies (e.g., SNOMED CT, LOINC) to describe data [20] [21].
- Common Data Elements (CDEs): Implementing CDEs across research teams and projects to ensure data is collected and structured consistently [22].
- Formal Semantics: Annotating data using formal semantics and common coordinate frameworks to ensure relationships between datasets are computationally accessible [22].
What is the difference between FAIR data and Open data?
FAIR and Open are distinct concepts. FAIR data is structured and described to be computationally actionable; it can be closed access, with strict security and permissions, yet still be Findable, Accessible, Interoperable, and Reusable by authorized users and systems [19]. Open data is defined by its lack of access restrictions and is made freely available to everyone. Not all open data is FAIR (e.g., a publicly available CSV file with no metadata), and not all FAIR data is open (e.g., a clinically sensitive genomic dataset in a secure, access-controlled repository) [19].
Troubleshooting Guides: Common FAIR Implementation Challenges
Issue 1: Data and Metadata Are Not Easily Discoverable
| Symptom | Possible Cause | Solution |
|---|---|---|
| Other researchers cannot locate your dataset. | Data is stored in personal or institutional storage without a persistent identifier. | Deposit data in a trusted repository that assigns a globally unique and persistent identifier (e.g., a DOI or Handle) [18] [20]. |
| Your dataset does not appear in relevant search engines. | Metadata is incomplete, uses non-standard terms, or is not registered in a searchable resource. | Create rich, machine-readable metadata using community-standardized schemas and ensure it is registered or indexed in a disciplinary resource [17] [20]. |
Issue 2: Inability to Integrate Multi-Omics Datasets
| Symptom | Possible Cause | Solution |
|---|---|---|
| Genomic and proteomic data from the same sample cannot be correlated. | Data formats are proprietary or inconsistent, and vocabularies are not aligned. | Use open, standard file formats (e.g., CSV, XML) and shared, broadly applicable ontologies (e.g., from the OBO Foundry) for all data and metadata [19] [20]. |
| Batch effects obscure biological signals when combining datasets from different labs. | A lack of harmonized protocols for sample preparation, data generation, and processing. | Implement and document Common Data Elements (CDEs) and standard operating procedures (SOPs) across all collaborating labs from the project's start [22]. |
Issue 3: Data Reuse is Hindered by Poor Provenance and Documentation
| Symptom | Possible Cause | Solution |
|---|---|---|
| You or others cannot replicate the analysis or understand the data's context. | Missing or unclear data usage license, provenance information, and methodological details. | Release data with a clear usage license and provide detailed provenance documentation that describes how the data was generated, processed, and analyzed [18] [20]. |
| The data's applicability for a new research question is uncertain. | Metadata lacks domain-relevant context and does not meet community standards. | Ensure metadata is richly described with a plurality of accurate attributes and is structured to meet domain-relevant community standards [20]. |
Experimental Protocols for FAIR Data Harmonization
Protocol: Implementing a Data Harmonization Framework for Team Science
Purpose: To establish a shared foundation for collecting, structuring, and sharing data within a large, interdisciplinary multi-omics consortium, enabling downstream integrated analyses [22].
Methodology:
- Establish Communication and Common Language: Facilitate workshops to build a shared vocabulary across computational and experimental researchers. This bridges disciplinary gaps and is the first step toward technical harmonization [22].
- Develop and Adopt Common Data Elements (CDEs): Collaboratively define the core set of data items that will be collected uniformly across all teams and experiments (e.g., standardized fields for sample ID, organism, tissue source, etc.) [22].
- Agree on Metadata Standards and Ontologies: Select and implement a minimal metadata standard specific to the project's data types (e.g., based on existing standards like the 3D Microscopy Metadata Standards). Mandate the use of agreed-upon controlled vocabularies and ontologies for semantic interoperability [22].
- Define the Data and Code Sharing Infrastructure: Select a common repository or platform with a defined dataset structure (e.g., the SPARC dataset structure) for publishing final, curated datasets. This ensures compliance with minimal metadata standards and facilitates discovery [22].
- Create a Data Management Plan (DMP): Document all agreed-upon standards, protocols, and infrastructure decisions in a DMP. This living document serves as the project's rulebook for FAIR data practices throughout the research lifecycle [20].
Workflow: The FAIRification Process for a Multi-Omics Dataset
The following diagram visualizes the pathway from raw, siloed data to a harmonized, FAIR-compliant dataset ready for integrated analysis.
| Tool Category | Example(s) | Function in FAIRification |
|---|---|---|
| Trusted Repositories | Zenodo, Figshare, Dataverse, Discipline-specific DBs [23] [20] | Provides a permanent home for data, assigns a Persistent Identifier (PID), and makes data discoverable and accessible. |
| Metadata Standards | ISA, SPARC Dataset Structure, 3D-MMS, CDISC [22] [20] [21] | Provides a structured schema for rich metadata collection, ensuring data is well-described and reusable. |
| Ontologies & Vocabularies | SNOMED CT, LOINC, OBO Foundry Ontologies [22] [21] | Provides standardized, machine-readable terms for data annotation, enabling semantic interoperability. |
| Data Formats | CSV, XML, JSON, RDF [20] | Open, non-proprietary formats ensure data can be read and processed by different computational systems in the long term. |
| Persistent Identifiers | Digital Object Identifier (DOI), Handle [18] [20] | A globally unique and permanent name for a dataset, making it reliably findable and citable. |
FAIR in Action: Multi-Omics Integration Workflow
The diagram below illustrates how FAIR principles enable the integration of disparate omics data layers through a unified computational analysis pipeline, leading to holistic biological insights.
The Critical Role of Rich Metadata and Standardized Ontologies
Frequently Asked Questions (FAQs) and Troubleshooting Guides
FAQ Category: Fundamentals of Metadata and Ontologies
Q1: What is the difference between data standardization and data harmonization? Standardization aims to unify data using a uniform methodology from the outset and can be seen as the most extreme form of stringent harmonization. Harmonization, however, is the practice of reconciling various types, levels, and sources of existing data into formats that are compatible and comparable for analysis [7]. It resolves heterogeneity in syntax (data format), structure (conceptual schema), and semantics (intended meaning) [7].
Q2: Why are minimum metadata requirements advocated over fixed standards in some areas of microbiome research? Due to the rapid technological progress in microbiome research, a flexible system that can be constantly improved is more practical than a rigid standard. Minimum requirements ensure essential information is captured while allowing for the evolution of new parameters as the field advances [24].
Q3: What are the core components of the FAIR principles that metadata should adhere to? Metadata should be curated to make data:
- Findable: Easy to locate by humans and computers.
- Accessible: Stored for long-term retrieval.
- Interoperable: Ready for integration with other data.
- Reusable: Fully described to allow replication and reuse [24].
FAQ Category: Implementation and Practical Challenges
Q4: I am preparing to submit my omics data to a public repository. What are the typical minimum metadata requirements? Common repositories often base their requirements on the MIxS (Minimum Information about any (x) Sequence) checklists [24]. While requirements can vary, the following table summarizes core elements often required:
| Metadata Category | Examples of Required Information |
|---|---|
| Investigation Details | Investigation type, project name [24] |
| Sample Details | Collection date, geographic location (latitude, longitude, country) [24] |
| Environmental Details | Biome, feature, material, selected environmental package [24] |
| Technical Methods | Sequencing method, library preparation protocols [24] |
Q5: A common error is the inconsistent use of ontologies, leading to data harmonization failures. How can I troubleshoot this?
- Problem: The same term is used for different concepts (e.g., "young adults" defined as 18-25 in one dataset and 18-30 in another).
- Solution: Implement a centralized reference ontology. For example, the OHDSI Standardized Vocabularies—which contains over 10 million concepts—provides a common framework that standardizes semantically equivalent concepts and supports international coding schemes, ensuring consistent meaning across datasets [25].
Q6: My multi-omics dataset has different data types with unique noise profiles and missing values. What is the first step to make them interoperable? The critical first step is preprocessing, which includes standardization and harmonization [15].
- Standardization: Ensures data is collected, processed, and stored consistently using agreed-upon protocols. This can involve normalizing data to account for differences in sample size, converting to a common scale, or removing technical biases [15].
- Harmonization: Aligns data from different sources by mapping them onto a common scale or reference, often using domain-specific ontologies [15].
FAQ Category: Advanced Data Integration and Integrity
Q7: What are the key challenges specific to multi-omics data integration? The table below outlines the primary challenges and their implications:
| Challenge | Description | Potential Consequence |
|---|---|---|
| Lack of Pre-processing Standards [2] | Each omics type (e.g., genomics, proteomics) has unique data structure, distribution, and batch effects. | Introduces variability, challenging harmonization. |
| Specialized Bioinformatics Expertise [2] | Requires cross-disciplinary knowledge in biostatistics, machine learning, and programming. | Major bottleneck in analysis. |
| Choice of Integration Method [2] | Multiple methods exist (e.g., MOFA, DIABLO, SNF), each with different approaches and outputs. | Confusion about the best method for a specific biological question. |
| Interpretation of Results [2] | Translating integrated outputs into actionable biological insight is complex. | Risk of drawing spurious conclusions. |
Q8: I've discovered a critical error in the metadata of a published dataset I am re-using. What should I do? Metadata integrity is a fundamental determinant of research credibility [26]. If you discover an error:
- Document the Error: Clearly identify the specific metadata field and the nature of the inaccuracy.
- Contact the Data Submitter: If contact information is available in the repository, reach out to them directly to alert them of the issue.
- Notify the Repository Curator: Submit a formal notice to the data repository (e.g., GEO, ENA) where the dataset is housed. They can place a note on the dataset record or contact the original submitters. Raising awareness of metadata errors is essential for maintaining the integrity of public data and preventing the propagation of incorrect findings [26].
Experimental Protocols and Workflows
Protocol 1: A Standardized Workflow for Multi-Omics Data Harmonization
This protocol provides a general methodology for harmonizing multi-omics data to ensure robustness and reproducibility.
Title: Multi-Omics Data Harmonization Workflow
Detailed Methodology:
- Data Preprocessing: Perform quality control, imputation of missing values, and noise reduction tailored to each omics data type (e.g., RNA-Seq, proteomics) [2].
- Standardization: Normalize data to account for differences in sample size or concentration. Convert data to a common scale or unit of measurement to ensure compatibility across platforms [15].
- Ontology Mapping: Map source data and metadata to a common, comprehensive reference ontology (e.g., OHDSI Standardized Vocabularies). This step standardizes semantically equivalent concepts and assigns domains according to clinical or biological categories [25].
- Data Harmonization: Resolve structural and semantic heterogeneity. This involves aligning data from different sources so they can be integrated, ensuring that the intended meaning of variables is consistent across all datasets [7].
- Apply Integration Method: Utilize a specific computational method (e.g., MOFA, DIABLO, SNF) to perform the integration based on the research question (supervised vs. unsupervised) [2].
Protocol 2: Implementing the FAIR Principles for Data Reusability
This protocol outlines key steps to make omics data Findable, Accessible, Interoperable, and Reusable.
Title: FAIR Data Principles Cycle
Detailed Methodology:
- Findable:
- Assign a persistent digital identifier (e.g., DOI) to your dataset.
- Describe the data with rich metadata, including the core elements from the MIxS checklist [24].
- Accessible:
- Deposit the data and metadata in a trusted, community-recognized repository (e.g., ENA, SRA, GEO).
- Ensure the data can be retrieved by their identifier using a standardized communication protocol.
- Interoperable:
- Use a formal, accessible, shared, and broadly applicable language for knowledge representation. This is achieved by using standardized ontologies and vocabularies (e.g., OHDSI, GO) [25].
- Qualify relationships between metadata elements using ontology terms.
- Reusable:
- Provide multiple, accurate, and relevant attributes to describe the data. Metadata should meet domain-specific community standards [24].
- Clearly state the license under which the data can be reused and associate detailed provenance information.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key resources for managing metadata and performing data harmonization in multi-omics studies.
| Tool / Resource Name | Type | Primary Function | Relevance to Data Harmonization |
|---|---|---|---|
| MIxS Checklists [24] | Reporting Standard | Defines minimum information for sequencing data. | Provides a common set of fields for describing genomic, metagenomic, and marker gene sequences, ensuring basic interoperability. |
| OHDSI Standardized Vocabularies [25] | Reference Ontology | A large-scale, centralized ontology for international health data. | Supports data harmonization by standardizing semantically equivalent concepts from over 136 source vocabularies, enabling cross-study analysis. |
| MOFA [2] | Integration Algorithm | Unsupervised factorization to infer latent factors from multi-omics data. | Discovers the principal sources of variation shared across different omics data modalities. |
| DIABLO [2] | Integration Algorithm | Supervised integration for biomarker discovery. | Integrates multiple omics datasets to find components that discriminate between known phenotypic groups. |
| SNF [2] | Integration Algorithm | Fuses sample similarity networks from different data types. | Constructs an overall integrated matrix capturing complementary information from all omics layers. |
| Omics Playground [2] | Analysis Platform | An all-in-one, code-free platform for multi-omics analysis. | Democratizes data integration by providing a cohesive interface with guided workflows and multiple state-of-the-art integration methods. |
Integration in Action: Strategic Frameworks and Analytical Techniques for Multi-Omics Data
FAQs on Multi-Omics Data Fusion
1. What are the main types of data fusion strategies, and how do they differ?
The three primary strategies for multi-omics data fusion are early, intermediate, and late fusion. Their core difference lies in the stage at which data from different omics layers are combined.
- Early Fusion (also known as data-level or feature-level fusion) involves concatenating raw or pre-processed features from each modality into a single, unified dataset before model training [27] [12].
- Intermediate Fusion (or joint fusion) integrates modality-specific features during the learning process itself, allowing the model to learn complex inter-modal relationships [28].
- Late Fusion (decision-level fusion) processes each modality through independent models and combines their predictions at the final decision stage [27] [29].
2. When should I choose late fusion over early fusion?
Late fusion is particularly advantageous when your dataset has a low sample-to-feature ratio, which is common in bioinformatics [29]. It is more robust to overfitting in scenarios with high-dimensional data (e.g., features on the order of 10⁵) and a limited number of patient samples (e.g., 10 to 10³) [29]. It also handles data heterogeneity effectively, as each modality can be processed with its own optimal pipeline [27] [29]. If your different omics data types have varying levels of informativeness or noise, late fusion allows the model to naturally weigh each modality based on its predictive power [29].
3. What are the common pitfalls of early fusion and how can they be mitigated?
The most significant pitfall of early fusion is the "curse of dimensionality", where concatenating features creates an extremely high-dimensional feature space that can lead to model overfitting, especially with small sample sizes [27] [12]. It also struggles with data heterogeneity, as different omics types may have unique data structures, scales, and noise profiles [29].
Mitigation strategies include:
- Applying robust dimensionality reduction (e.g., PCA, autoencoders) before concatenation [29].
- Using strong regularization techniques in the subsequent model to prevent overfitting [29].
- Ensuring careful data normalization and harmonization across all modalities to make features more compatible [12].
4. How does intermediate fusion capture relationships between omics layers?
Unlike early and late fusion, intermediate fusion uses specialized model architectures that allow interaction between modalities during feature learning [28]. Techniques such as attention mechanisms can learn to weight the importance of specific features from different omics [27], while neural networks with shared layers can learn a joint representation that captures non-linear dependencies between, for instance, gene expression and protein abundance data [28]. This often leads to more biologically insightful models [28].
5. Is there a one-size-fits-all best fusion strategy?
No, the optimal fusion strategy is highly problem-specific and data-dependent [29]. The best choice depends on factors like sample size, data dimensionality, heterogeneity, and the specific biological question. Research indicates that late fusion often outperforms others in classical bioinformatics settings with limited samples and high-dimensional features [29], whereas early or intermediate fusion may be more effective in scenarios with larger sample sizes and fewer total features [29].
Comparison of Fusion Strategies
Table 1: Advantages and challenges of different multi-omics integration strategies.
| Strategy | Description | Advantages | Challenges |
|---|---|---|---|
| Early Fusion | Raw or pre-processed features from all omics are combined into a single input vector [27] [12]. | Simplicity of implementation; potential to capture all cross-omics interactions [12]. | High risk of overfitting with small sample sizes; requires all modalities to be present for each sample [27] [29]. |
| Intermediate Fusion | Data is integrated during model training, often using specialized architectures [28]. | Can capture complex, non-linear relationships between omics layers [27] [28]. | Increased model complexity; can be computationally intensive [28]. |
| Late Fusion | Separate models are built for each omics type, and their predictions are combined [27] [29]. | Robustness to overfitting and missing data; allows modality-specific preprocessing [27] [29]. | May miss subtle cross-omics interactions [12]. |
Table 2: Guide to selecting a fusion strategy based on data characteristics and research objectives.
| Criterion | Recommended Strategy | Rationale |
|---|---|---|
| Small Sample Size (n) & High Dimensionality (p) | Late Fusion | Reduces overfitting risk by building simpler, modality-specific models [29]. |
| Large Sample Size & Lower Dimensionality | Early or Intermediate Fusion | Sufficient data is available to learn complex, cross-modal patterns without overfitting [29]. |
| Primary Goal: Robust Prediction | Late Fusion | Proven to provide higher accuracy and robustness in survival prediction for cancer patients [29]. |
| Primary Goal: Biological Insight | Intermediate Fusion | Can reveal how different omics layers interact, providing mechanistic understanding [28]. |
| Presence of Missing Modalities | Late Fusion | Individual models can be trained on available data, and predictions are combined afterward [12]. |
Experimental Protocols
Protocol 1: Implementing a Late Fusion Workflow for Survival Prediction
This protocol is based on a machine learning pipeline that demonstrated consistent outperformance of single-modality approaches in cancer survival prediction using TCGA data [29].
1. Data Preprocessing and Dimensionality Reduction per Modality:
- Input: Separate datasets for each omics modality (e.g., transcripts, proteins, metabolites, clinical data) [29].
- Normalization: Apply modality-specific normalization (e.g., TPM for RNA-seq, intensity normalization for proteomics) [12].
- Feature Selection: For each modality, reduce dimensionality using supervised feature selection. In the referenced study, linear or monotonic methods (e.g., Pearson or Spearman correlation with the target) outperformed non-linear methods in this context [29].
- Output: A reduced, informative feature set for each omic type.
2. Train Unimodal Survival Models:
- For each processed omics modality, train an independent predictive model. The referenced pipeline found that ensemble methods like gradient boosting or random forests can be effective [29].
- Validate each model's performance rigorously using multiple training-test splits and report confidence intervals for metrics like the C-index [29].
3. Fuse Predictions:
- Combine the predictions (e.g., risk scores) from each unimodal model into a final ensemble prediction.
- Use a simple averaging or a weighted averaging scheme, where weights can be based on the unimodal model's performance [29] [12].
Protocol 2: An Intermediate Fusion Approach Using a Neural Network
This protocol outlines the steps for using a neural network to learn joint representations of multi-omics data, suitable for tasks like subtype classification [28].
1. Input Stream Setup:
- Design separate input branches for each omics data type (e.g., genomics, transcriptomics, proteomics). Each branch should accept a feature vector from its respective modality [28].
2. Feature Learning and Compression:
- Each input branch can consist of one or more fully connected layers that act as a modality-specific encoder. The goal is to transform the raw input into a meaningful representation [28].
- Alternatively, use a method like a Variational Autoencoder (VAE) per modality to compress the data into a lower-dimensional latent space [12].
3. Representation Fusion and Model Training:
- Concatenate the outputs (the learned features) from all modality-specific branches. This concatenated vector forms the joint representation [28].
- Feed this joint representation into a final set of fully connected layers to perform the prediction task (e.g., classification or regression) [28].
- Train the entire network (all branches and the joint head) end-to-end, allowing the model to learn which cross-modal features are most relevant for the task [28].
Multi-Omics Fusion Workflows
The Scientist's Toolkit
Table 3: Essential computational tools and reagents for multi-omics data fusion.
| Tool / Reagent | Type | Primary Function | Example Use Case |
|---|---|---|---|
| Seurat [30] | Software Tool | Weighted nearest-neighbor integration for single-cell multi-omics data. | Integrating mRNA expression and chromatin accessibility data from the same cell [30]. |
| MOFA+ [30] | Software Tool | Factor analysis-based integration to disentangle variation across omics layers. | Identifying common sources of variation in unmatched multi-omics datasets (e.g., mRNA, DNA methylation) [30]. |
| GLUE (Graph-Linked Unified Embedding) [30] | Software Tool | Variational autoencoder that uses prior biological knowledge to anchor features for integration. | Triple-omic integration of chromatin accessibility, DNA methylation, and mRNA data [30]. |
| The Cancer Genome Atlas (TCGA) [11] | Data Repository | Provides large-scale, publicly available multi-omics datasets (genomics, epigenomics, transcriptomics, proteomics) from cancer patients. | Benchmarking and training multi-omics fusion models for cancer subtype classification or survival prediction [11]. |
| Autoencoders (AEs) / Variational Autoencoders (VAEs) [12] | ML Method | Neural networks for non-linear dimensionality reduction, creating a lower-dimensional latent representation of high-dimensional omics data. | Compressing transcriptomics and proteomics data into a shared latent space for intermediate fusion [12]. |
Leveraging AI and Machine Learning for Pattern Recognition and Data Fusion
Frequently Asked Questions (FAQs)
Q1: What are the most significant data-related challenges when beginning a multi-omics study? The primary challenges, often called the "four Vs" of big data, are Volume (high-dimensional data where features far exceed samples), Variety (structural differences between data types like discrete mutations vs. continuous protein measurements), Velocity (managing real-time data streams), and Veracity (distinguishing biological signals from technical noise and batch effects) [31]. Computational scalability and the "curse of dimensionality" are also major hurdles [31].
Q2: Which AI models are best suited for integrating disparate omics data types? No single model is best for all scenarios, but several have proven effective [31] [32] [11]:
- Graph Neural Networks (GNNs) are ideal for modeling known biological structures, such as protein-protein interaction networks perturbed by mutations [31].
- Multi-modal Transformers excel at fusing fundamentally different data types, such as MRI radiomics with transcriptomic data [31].
- Fully Connected Neural Networks (FCNs), especially when enhanced with contrastive learning and domain-specific embeddings (e.g., BioBERT), are highly effective for harmonizing metadata and variable descriptions across cohort studies [32].
- Convolutional Neural Networks (CNNs) are used for image-based data, such as automatically quantifying protein staining in tissue samples with pathologist-level accuracy [31].
Q3: How can I handle missing data in one or more omics layers? Advanced imputation strategies are recommended over simply removing features or samples. Matrix factorization and deep learning (DL)-based reconstruction methods can intelligently estimate missing values based on patterns in the available data [31]. The pervasive nature of missing data due to technical limitations makes this a critical step in the preprocessing workflow [31].
Q4: What does "data harmonization" mean in this context, and can it be automated? Data harmonization is the process of standardizing disparate variables and metadata across multiple datasets into a unified format [32]. This is crucial for cross-study analysis. Yes, it can be automated using Natural Language Processing (NLP). For example, one method uses a Fully Connected Neural Network with BioBERT embeddings to classify variable descriptions from different studies (e.g., "SystolicBP" vs. "SBPvisit1") into unified medical concepts with high accuracy (AUC of 0.99) [32].
Q5: Why are my AI models performing well on training data but failing to generalize to new datasets? This is often due to batch effects—technical variations introduced by different sequencing platforms, laboratories, or protocols. To improve generalizability, employ rigorous batch correction tools like ComBat and ensure your model validation includes external validation on a completely independent dataset [31]. Techniques like federated learning also allow for model training across institutions without sharing raw data, which can improve robustness [31].
Troubleshooting Guides
Issue 1: Poor Model Performance Due to Technical Batch Effects
Problem: Your model's predictive accuracy drops significantly when applied to data generated from a different site or platform.
Solution: Implement a rigorous batch correction and validation pipeline.
- Step 1: Diagnose Batch Effects. Use Principal Component Analysis (PCA) or other visualization tools to see if samples cluster more strongly by batch (e.g., lab ID) than by biological condition.
- Step 2: Apply Batch Correction. Utilize tools like ComBat or other normalization methods to remove technical artifacts while preserving biological signals [31].
- Step 3: Validate Externally. Always test the final model on an external cohort that was not used in any part of the training or tuning process [31]. This is the gold standard for assessing true generalizability.
Issue 2: Inability to Integrate Heterogeneous Data Types
Problem: You have genomic, proteomic, and image data, but cannot effectively fuse them into a single analytical framework.
Solution: Choose an integration method based on your scientific objective. The table below summarizes the main approaches.
Table 1: Multi-Omics Data Integration Methods and Tools
| Scientific Objective | Description | Example Methods | Reference |
|---|---|---|---|
| Subtype Identification | Discover novel disease subtypes by grouping patients based on multi-omics profiles. | Clustering (e.g., iCluster), Matrix Factorization | [11] |
| Detect Disease-Associated Patterns | Identify complex molecular patterns and biomarkers correlated with a condition. | Multi-Kernel Learning, Pattern Recognition | [11] |
| Understand Regulatory Processes | Uncover how changes at one molecular level (e.g., epigenomics) affect another (e.g., transcriptomics). | Network Inference (e.g., GNNs), Bayesian Networks | [31] [11] |
| Diagnosis/Prognosis | Build classifiers to predict patient outcome or disease state. | Supervised ML/DL (e.g., Transformers, CNNs) | [31] [11] |
| Drug Response Prediction | Predict a patient's sensitivity or resistance to a specific therapy. | Regression Models, "Digital Twin" simulations | [31] |
Issue 3: The "Black Box" Problem – Lack of Model Interpretability
Problem: Your model makes accurate predictions, but you cannot understand how it arrived at them, which is critical for biological insight and clinical trust.
Solution: Integrate Explainable AI (XAI) techniques into your workflow.
- Step 1: Use Inherently Interpretable Models. For simpler tasks, start with models like decision trees or logistic regression, which are more transparent.
- Step 2: Apply Post-Hoc Explanation Methods. For complex models like deep neural networks, use techniques such as SHapley Additive exPlanations (SHAP). SHAP quantifies the contribution of each input feature (e.g., a specific gene mutation) to the final prediction, making the model's decision process clearer [31].
- Step 3: Biological Validation. Use the feature importance scores from XAI to prioritize findings (e.g., key genes or pathways) for downstream experimental validation in the lab.
Experimental Protocols
Protocol 1: NLP-Based Automated Data Harmonization
This protocol details the method for using a Fully Connected Neural Network (FCN) to harmonize variable metadata, as described in [32].
1. Objective: To automatically map free-text variable names and descriptions from different biomedical datasets into harmonized medical concepts.
2. Materials & Reagents:
- Datasets: Metadata (variable names and descriptions) from cohort studies (e.g., ARIC, MESA, FHS).
- Pretrained Language Model: BioBERT (Bidirectional Encoder Representations from Transformers for Biomedical Text Mining).
- Computing Environment: Standard deep learning framework (e.g., PyTorch, TensorFlow).
3. Procedure:
- Step 1 - Data Preparation: Extract all variable descriptions. Manually annotate a subset into predefined harmonized concepts (e.g., "systolic blood pressure," "diabetes medication") to create a labeled ground truth.
- Step 2 - Generate Embeddings: Convert each variable description into a 768-dimensional semantic vector using the pretrained BioBERT model.
- Step 3 - Create Paired Dataset: Frame the task as a binary classification. Generate pairs of variable descriptions and label them as either belonging to the same concept (matched pair) or not (non-matched pair). Maintain a balanced ratio (e.g., 1:3) of matched to non-matched pairs.
- Step 4 - Model Training: Train an FCN classifier. The input is the cosine similarity between the BioBERT embedding vectors of a paired description. The network uses binary cross-entropy loss and the Adam optimizer.
- Step 5 - Inference: For a new, unlabeled variable description, the model calculates its similarity to all known concept representatives and assigns it to the concept with the highest similarity score.
4. Expected Results: The published FCN model achieved a top-5 accuracy of 98.95% and an Area Under the Curve (AUC) of 0.99, significantly outperforming a logistic regression baseline (AUC 0.82) [32].
Diagram 1: NLP-based data harmonization workflow.
Protocol 2: AI-Driven Multi-Omics Integration for Patient Subtyping
1. Objective: To integrate genomic, transcriptomic, and proteomic data to identify novel, clinically relevant disease subtypes.
2. Materials & Reagents:
- Omics Data: Matched genomic (SNVs/CNVs), transcriptomic (RNA-seq), and proteomic (mass spectrometry) data from the same patient cohort.
- Data Repositories: Publicly available data from sources like The Cancer Genome Atlas (TCGA) [11].
- Computational Tools: Cloud-based analytics platforms (e.g., AWS with SageMaker, HealthOmics, Athena) or local high-performance computing clusters [33].
3. Procedure:
- Step 1 - Data Preprocessing & Harmonization: Independently preprocess each omics layer. This includes quality control, normalization (e.g., DESeq2 for RNA-seq), and batch effect correction (e.g., using ComBat) [31].
- Step 2 - Dimensionality Reduction: Apply feature selection or extraction (e.g., PCA) to each data modality to reduce noise and computational complexity.
- Step 3 - Data Integration: Use an intermediate integration method designed for subtype identification. A common approach is multi-omics matrix factorization, which learns a joint representation of the patient across all data types in a lower-dimensional space.
- Step 4 - Clustering: Apply a clustering algorithm (e.g., k-means, hierarchical clustering) on the integrated patient representations to identify distinct molecular subtypes.
- Step 5 - Clinical Validation: Correlate the identified subtypes with clinical outcomes (e.g., overall survival, response to therapy) to assess their biological and clinical relevance.
4. Expected Results: Discovery of patient subgroups with distinct multi-omics profiles and significantly different survival outcomes, which may not be identifiable using single-omics data alone. For example, one study reported integrated classifiers with AUCs between 0.81–0.87 for early-detection tasks [31].
Diagram 2: Multi-omics integration and subtyping workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for AI-Driven Multi-Omics Research
| Tool / Resource Name | Type | Primary Function in Multi-Omics | Reference / Link |
|---|---|---|---|
| BioBERT | Pretrained Language Model | Generates domain-specific semantic embeddings for biomedical text, enabling automated metadata harmonization. | [32] |
| ComBat | Statistical Algorithm | Removes batch effects from high-dimensional datasets to improve data quality and model generalizability. | [31] |
| SHAP (SHapley Additive exPlanations) | Explainable AI (XAI) Library | Interprets complex AI model outputs by quantifying the contribution of each feature to a prediction. | [31] |
| Graph Neural Networks (GNNs) | AI Model Architecture | Models biological networks (e.g., protein-protein interactions) to uncover dysregulated pathways. | [31] |
| The Cancer Genome Atlas (TCGA) | Data Repository | Provides curated, publicly available multi-omics datasets from cancer patients for analysis and benchmarking. | [11] |
| AWS HealthOmics & SageMaker | Cloud Computing Platform | Offers managed services for storing, processing, and analyzing multi-omics data at scale. | [33] |
| Multi-Kernel Learning | Data Integration Method | Fuses different omics data types by assigning each a separate "kernel" function, then combining them. | [11] |
FAQs and Troubleshooting Guides
This section addresses common challenges researchers face during data pre-processing for multi-omics studies, providing targeted solutions and best practices.
FAQ 1: How should I handle missing data in my multi-omics dataset before running machine learning models?
- Problem: Machine learning models often fail or perform poorly when faced with missing values, which are pervasive in real-world omics data [34] [35].
- Solutions:
- Do not simply ignore missing values. Most algorithms cannot handle them and will produce errors [34].
- Use imputation. Replacing missing values with plausible estimates is the standard approach. The best method depends on your data and the missingness mechanism [36] [35].
- Impute before feature selection. Research indicates that performing imputation before feature selection leads to better model performance, as measured by recall, precision, F1-score, and accuracy [35].
- Troubleshooting: If your model's performance is poor after imputation, investigate the pattern of missingness (e.g., MCAR, MAR, NMAR) and try a more advanced imputation method. Simple methods like mean imputation can distort data distribution and variance [35].
FAQ 2: My data comes from different experimental batches. How can I correct for technical batch effects without removing true biological signals?
- Problem: Batch effects are technical biases from different library preps, sequencing runs, or sample handling that can obscure real biology and generate false signals [37].
- Solutions:
- Use established correction methods. Algorithms like ComBat and limma are designed to model and remove batch effects while preserving biological variation [38].
- Consider data incompleteness. For omic data with many missing values, standard tools may fail. Use methods specifically designed for incomplete data, such as Batch-Effect Reduction Trees (BERT) or HarmonizR, which retain more numeric values during integration [38].
- Leverage covariates and references. When batch designs are imbalanced, specify categorical covariates (e.g., biological conditions) or use reference samples to guide the correction process for more robust results [38].
- Troubleshooting: After correction, always validate that known biological signals persist. There is a risk of both under-correction (leaving residual bias) and over-correction (removing true biological variation) [37].
FAQ 3: What is the difference between data normalization for databases and for machine learning?
- Problem: The term "normalization" is used in two distinct contexts, which can cause confusion.
- Solutions:
- Database Normalization: This is a structural process for organizing data in a relational database to reduce redundancy and improve integrity. It follows rules called "normal forms" (1NF, 2NF, 3NF) [39] [40].
- Machine Learning Normalization (Feature Scaling): This is a mathematical process of bringing numeric features to a common scale to prevent algorithms with distance-based calculations from being skewed by the original magnitude of the features [39] [34].
- Troubleshooting: For machine learning, if your model is converging slowly or is dominated by a few features, you likely need to apply feature scaling (e.g., standardization, normalization) to your numerical data [34].
FAQ 4: Should I perform imputation before or after normalizing or correcting batch effects in a multi-omics workflow?
- Problem: The order of operations in a pre-processing pipeline can significantly impact the final results.
- Solution: The recommended workflow is to correct for batch effects before performing any other normalization or imputation steps [38] [41]. Batch effect correction methods are designed to handle incomplete data, and applying them first ensures that subsequent steps like data-specific normalization are not confounded by technical noise. Imputation can then be performed on the batch-corrected data.
Data Pre-processing Method Comparisons
The tables below summarize key quantitative findings and methodologies from recent research to guide your experimental design.
Table 1: Benchmarking of Missing Data Imputation Techniques on Healthcare Diagnostic Datasets [35]
| Imputation Technique | Description | Key Finding (RMSE/MAE) |
|---|---|---|
| MissForest | Uses a Random Forest model to predict missing values iteratively. | Best performance on tested healthcare datasets. |
| MICE | Generates multiple imputations using chained equations. | Second-best performance after MissForest. |
| KNN Imputation | Fills missing values by averaging the k-nearest neighbors. | Robust and effective, but performance varies. |
| Interpolation | Fills values using linear interpolation between points. | Outperformed mean imputation in environmental data [35]. |
| Mean/Median Imputation | Replaces missing values with the feature's mean or median. | Simple but can distort variable distribution and variance. |
| LOCF | Carries the last observation forward. | Common in clinical research; assumes value stability. |
Table 2: Evaluation of Normalization Methods for Mass Spectrometry-Based Multi-Omics Data in a Temporal Study [41]
| Normalization Method | Core Assumption | Recommended For |
|---|---|---|
| Probabilistic Quotient (PQN) | The overall distribution of feature intensities is similar across samples. | Metabolomics, Lipidomics, Proteomics |
| LOESS (with QC samples) | The proportion of up- and down-regulated features is balanced. | Metabolomics, Lipidomics, Proteomics |
| Median Normalization | The median feature intensity is constant across samples. | Proteomics |
| SERRF | Machine learning method using QC samples to correct systematic errors. | Can outperform others in metabolomics but may mask biological variance. |
Table 3: Data Integration Tools for Incomplete Omic Data with Batch Effects [38]
| Tool / Method | Approach | Key Advantage |
|---|---|---|
| BERT | Tree-based framework using ComBat/limma for pairwise batch correction. | Retains all numeric values; fast; handles covariate imbalance. |
| HarmonizR | Matrix dissection to create complete sub-matrices for parallel integration. | The first method to handle arbitrarily incomplete data. |
| Standard ComBat/limma | Empirical Bayes methods for batch-effect correction. | Established methods, but require complete data matrices. |
Experimental Protocols
Protocol 1: Evaluating Imputation Techniques for Healthcare Data
This protocol is adapted from a 2025 comparative study [35].
- Dataset Selection: Acquire relevant, complete healthcare datasets (e.g., breast cancer, heart disease, PIMA diabetes from public repositories).
- Introduce Missingness: To simulate a real-world scenario, artificially introduce missing values (e.g., at 10%, 15%, 20%, 25%) under the Missing Completely at Random (MCAR) mechanism.
- Apply Imputation Methods: Implement a suite of imputation techniques, including:
- Mean Imputation
- Median Imputation
- Last Observation Carried Forward (LOCF)
- K-Nearest Neighbor (KNN) Imputation
- Linear Interpolation
- MissForest
- Multiple Imputation by Chained Equations (MICE)
- Evaluate Performance: Compare the methods using error metrics such as Root Mean Squared Error (RMSE) and Mean Absolute Error (MAE) by comparing the imputed values to the known, original values.
- Downstream Analysis: To test the impact on machine learning, perform feature selection and classification after imputation and evaluate using recall, precision, F1-score, and accuracy.
Protocol 2: Assessing Normalization Strategies for Multi-Omics Time-Course Data
This protocol is based on a 2025 evaluation of mass spectrometry normalization strategies [41].
- Data Generation: Generate metabolomics, lipidomics, and proteomics datasets from the same biological sample lysates to control for biological variability.
- Data Preparation: Process raw data using standard software (e.g., Compound Discoverer for metabolomics, MS-DIAL for lipidomics, Proteome Discoverer for proteomics). Perform initial filtering and gap-filling.
- Apply Normalization: Apply a range of normalization methods to each omics dataset, such as:
- Total Ion Current (TIC)
- Median Normalization
- Probabilistic Quotient Normalization (PQN)
- LOESS (with and without QC samples)
- Quantile Normalization
- Variance Stabilizing Normalization (VSN)
- SERRF
- Evaluate Effectiveness: Assess the normalization based on two criteria:
- Technical Improvement: Check the consistency of features in Quality Control (QC) samples. Good normalization improves QC consistency.
- Biological Preservation: Analyze the variance in the data. A good method should reduce technical variance while preserving or enhancing variance explained by time and treatment factors.
The Scientist's Toolkit
Table 4: Essential Research Reagents and Computational Tools
| Item | Function in Pre-processing | Example / Note |
|---|---|---|
| Pooled QC Samples | A quality control sample made by mixing aliquots of all study samples. Used by normalization methods (e.g., LOESS, SERRF) to model and correct technical variation across a run [41]. | Critical for mass spectrometry-based omics. |
| Python Packages | Provide libraries for implementing imputation and scaling. | imputena & missingpy for imputation [35]; pandas & scikit-learn for general preprocessing [34]. |
| R/Bioconductor Packages | Provide statistical methods for batch effect correction and normalization. | limma, ComBat for batch correction [38]; vsn for normalization [41]. |
| BERT (Software) | A high-performance R tool for batch-effect reduction on incomplete omic profiles. Retains more data and handles complex covariates compared to earlier tools [38]. | Available on Bioconductor. |
| Pluto Bio Platform | A commercial, no-code platform designed for multi-omics data harmonization and visualization, simplifying batch effect correction for non-bioinformaticians [37]. |
Workflow and Relationship Diagrams
This diagram illustrates the logical workflow for pre-processing multi-omics data, integrating the key steps discussed in the FAQs and protocols.
Recommended Multi-Omics Pre-processing Workflow
This diagram visualizes the core-branch structure of the Batch-Effect Reduction Trees (BERT) algorithm, which efficiently integrates incomplete datasets.
BERT Algorithm Core-Branch Structure
Network integration is a powerful computational approach that addresses a central challenge in modern biomedical research: how to meaningfully combine multiple layers of biological information. This method involves mapping various omics datasets—genomics, transcriptomics, proteomics, and metabolomics—onto shared biochemical networks to improve mechanistic understanding of disease processes [5]. Unlike simpler integration methods that might only correlate findings from separate analyses, network integration interweaves multiple omics profiles into a single dataset for higher-level analysis, where analytes are connected based on known interactions [5]. This approach allows researchers to pinpoint biological dysregulation to single reactions, enabling the identification of actionable therapeutic targets that might remain hidden when examining individual omics layers in isolation.
The foundational principle of network integration rests on representing biological knowledge as structured networks. In these networks, nodes represent biological entities such as genes, transcripts, proteins, and metabolites, while edges represent the known functional or physical interactions between them [2]. For example, a transcription factor can be connected to the transcript it regulates, or metabolic enzymes can be linked to their associated metabolite substrates and products [5]. By mapping experimental multi-omics data onto these predefined networks, researchers can identify dysregulated pathways and modules that span multiple biological layers, offering a systems-level perspective on health and disease that is essential for advancing precision medicine [12].
Key Methods for Multi-Omics Network Integration
Similarity Network Fusion (SNF)
Similarity Network Fusion (SNF) constructs and fuses patient-similarity networks to create a comprehensive view of biological systems. Rather than merging raw measurements directly, SNF creates a separate sample-similarity network for each omics dataset, where nodes represent patients or biological specimens and edges encode the similarity between samples based on that specific data type [2]. These data type-specific matrices are then fused through a non-linear process that strengthens strong similarities and removes weak ones across omics layers, generating a unified network that captures complementary information from all modalities [12] [2].
This method is particularly powerful for disease subtyping, as the fused network can reveal patient subgroups that might not be apparent when analyzing any single omics dataset. The iterative fusion process enables SNF to effectively handle different data types with varying scales and distributions, making it robust for integrating diverse omics measurements. The resulting fused network serves as a foundation for further analyses, including clustering to identify disease subtypes or prognostic groups that consider the full complexity of multi-omics profiles [12].
Network-Based Integration Using Biochemical Knowledge
Network-based integration methods utilize existing biochemical knowledge to create a framework for integrating multi-omics data. This approach first transforms each omics dataset into a biological network representation, such as gene co-expression networks or protein-protein interaction networks [12]. These networks are then integrated to reveal functional relationships and modules that drive disease processes.
The core strength of this approach lies in its incorporation of established biological context through networks. For example, researchers can map multi-omics data onto shared biochemical networks where multiple omics datasets are connected based on known interactions [5]. This might include connecting transcription factors to their target genes, metabolic enzymes to their substrates and products, or proteins to their functional partners in protein complexes [5]. By using these established relationships as scaffolding for integration, this method ensures that resulting models reflect biologically plausible mechanisms rather than just statistical correlations.
Graph Convolutional Networks (GCNs) represent a sophisticated implementation of this approach, where deep learning algorithms operate directly on network-structured biological data [12]. GCNs learn from network topology by aggregating information from a node's neighbors to make predictions, effectively propagating information across the network to identify functionally relevant patterns in multi-omics data [12].
Table 1: Comparison of Network Integration Methods
| Method | Primary Approach | Key Advantages | Common Applications |
|---|---|---|---|
| Similarity Network Fusion (SNF) | Fuses patient-similarity networks from each omics layer | Robust to noise; handles different data types effectively; non-linear integration | Disease subtyping; prognosis prediction; patient stratification |
| Network-Based Integration | Maps omics data onto known biological networks | Incorporates prior biological knowledge; results are more interpretable | Identifying dysregulated pathways; mechanistic insights; biomarker discovery |
| Graph Convolutional Networks (GCNs) | Deep learning on graph-structured biological data | Learns complex patterns from network topology; powerful predictive capability | Clinical outcome prediction; drug response prediction; feature learning |
Frequently Asked Questions (FAQs)
Q1: What are the primary technical challenges when implementing network integration for multi-omics data?
The main challenges include data heterogeneity, where each omics layer has different formats, scales, and statistical distributions [2] [42]; batch effects introduced by technical variations across different processing batches [12]; missing data points that are common in proteomics and metabolomics datasets [42]; and the computational complexity of analyzing high-dimensional data [12]. Additionally, ID conversion—correlating identities of the same biological entities across multiple omics layers—presents significant difficulties, as different databases may use inconsistent nomenclature [42].
Q2: How can researchers address the problem of data heterogeneity in network integration?
Data normalization and harmonization are essential first steps. Each omics data type requires tailored preprocessing, including normalization to make measurements comparable across platforms [12]. For RNA-seq data, this might include TPM or FPKM normalization, while proteomics data requires intensity normalization [12]. Additionally, specialized statistical methods like ComBat can remove batch effects, and robust imputation methods such as k-nearest neighbors (k-NN) or matrix factorization can address missing data issues [12]. Establishing standardized preprocessing protocols for each data type before integration is critical for success.
Q3: What are the sample preparation requirements for multi-omics studies aiming for network integration?
For optimal network integration, multi-omics profiles should ideally be acquired concurrently from the same set of samples (matched multi-omics) rather than different, unpaired samples [2]. This maintains biological context and enables more refined associations between molecular modalities. For single-cell multi-omics approaches, nuclear integrity is paramount—nuclear membranes should show well-resolved edges without blebbing or disintegration [43]. For tissue samples, proper preservation in liquid nitrogen (not -80°C) is recommended, and nuclei samples should be used immediately rather than preserved [43].
Q4: How do I choose between different network integration methods for my specific research question?
Method selection should be guided by your research objective. For disease subtyping, SNF has proven effective [2]. For understanding regulatory mechanisms and pathway dysregulation, knowledge-based network integration is preferable [5]. If you have a specific prediction task such as clinical outcome or drug response, Graph Convolutional Networks may be most appropriate [12]. Consider whether your approach requires unsupervised pattern discovery (use SNF) or supervised prediction (use GCNs), and the availability of well-annotated biological networks for your system of interest.
Troubleshooting Common Experimental Issues
Problem: Inconsistent Results Across Omics Layers
Issue: Molecular patterns observed in one omics layer do not correspond to expectations in another layer.
Solution:
- Verify sample quality: Ensure that all omics measurements come from the same biological sample and that degradation has not occurred. For single-cell studies, assess nuclear membrane integrity under a microscope [43].
- Check normalization methods: Apply appropriate normalization for each data type before integration. RNA-seq data may require TPM or FPKM normalization, while proteomics needs intensity normalization [12].
- Consider biological timing: Account for potential time delays between molecular events. For example, mRNA release and protein production may not be temporally aligned [44].
- Apply correlation analysis: Use Pearson's or Spearman's correlation to assess transcription-protein correspondence before network integration [44].
Problem: Poor Quality Networks After Integration
Issue: Integrated networks are too dense or too sparse, making biological interpretation difficult.
Solution:
- Optimize similarity thresholds: In SNF, adjust the number of neighbors and scaling parameters to control network density [2].
- Implement filtering strategies: Apply correlation thresholds based on R² values and p-values to retain only meaningful connections [44].
- Use community detection: Apply multilevel community detection algorithms to identify functionally relevant modules within complex networks [44].
- Incorporate prior knowledge: Constrain networks using established biological interactions from databases to improve biological relevance [5].
Diagram 1: Troubleshooting workflow for poor quality network integration
Problem: Computational Limitations with Large Multi-Omics Datasets
Issue: Network integration algorithms become computationally intractable with large sample sizes or feature numbers.
Solution:
- Dimensionality reduction: Apply feature selection before integration using methods like MOFA or autoencoders to compress high-dimensional data into lower-dimensional latent spaces [12].
- Cloud computing: Utilize scalable cloud infrastructure and distributed computing frameworks to handle petabyte-scale data [12].
- Subsampling strategies: Implement strategic sampling approaches that maintain biological diversity while reducing computational burden.
- Algorithm optimization: Choose methods with computational complexity appropriate for your dataset size, considering approximations when exact solutions are infeasible.
Experimental Protocols for Network Integration
Protocol: Similarity Network Fusion for Disease Subtyping
Purpose: To identify disease subtypes by integrating multiple omics datasets using Similarity Network Fusion.
Materials Needed:
- Normalized multi-omics datasets from the same patient samples
- Computational environment with SNF software installed
- Clinical metadata for result validation
Procedure:
- Data Preprocessing: Normalize each omics dataset separately using appropriate methods for each data type.
- Similarity Matrix Construction: For each omics data type, construct a patient similarity matrix using an appropriate distance metric.
- Network Fusion: Iteratively fuse the similarity matrices using SNF algorithm to create a unified patient network.
- Clustering Analysis: Apply spectral clustering to the fused network to identify patient subgroups.
- Validation: Compare identified subtypes with clinical outcomes to assess biological relevance.
Troubleshooting Tips:
- If clusters do not separate clearly, adjust the number of neighbors parameter in SNF.
- If results are driven by one data type, check for batch effects in individual omics datasets.
- Validate findings using external datasets or functional enrichment analysis.
Protocol: Knowledge-Based Network Integration for Pathway Analysis
Purpose: To map multi-omics data onto established biological pathways to identify dysregulated mechanisms.
Materials Needed:
- Multi-omics datasets with consistent sample identifiers
- Reference pathway databases
- Network analysis software
Procedure:
- Network Preparation: Select appropriate biological networks relevant to your research question.
- Data Mapping: Project each omics dataset onto the network structure.
- Integration: Overlay multiple omics layers onto the same network nodes and edges.
- Module Detection: Identify network modules that show coordinated changes across omics layers.
- Functional Interpretation: Perform enrichment analysis on identified modules.
Table 2: Essential Research Reagents and Computational Tools for Network Integration
| Resource Type | Specific Examples | Function/Purpose |
|---|---|---|
| Software Tools | SNF, MOFA, DIABLO, xMWAS | Implement specific network integration algorithms |
| Biological Networks | Protein-protein interactions, metabolic pathways, gene regulatory networks | Provide scaffolding for data integration |
| Reference Databases | KEGG, Reactome, GO, STRING | Source of established biological interactions |
| Programming Environments | R, Python with specialized packages | Data preprocessing, analysis, and visualization |
| Visualization Tools | Cytoscape, Gephi | Visual exploration and interpretation of integrated networks |
Advanced Applications and Future Directions
Network integration of multi-omics data is increasingly being applied in translational research contexts. In oncology, this approach has been used to identify distinct molecular subtypes of cancers that respond differently to treatments [5]. For complex diseases, network integration helps unravel the interplay between genetic predisposition and environmental factors by connecting genomic variants to their functional consequences across multiple molecular layers [12]. The approach is particularly powerful for biomarker discovery, as it can identify multi-omics signatures that are more robust than single-layer biomarkers [2].
Emerging methodologies in network integration include the incorporation of artificial intelligence and machine learning techniques to enhance pattern recognition in complex biological networks [5]. Graph neural networks represent a particularly promising direction, as they can learn directly from network-structured data while incorporating multiple types of biological relationships [12]. Additionally, approaches that combine both data-driven and knowledge-driven elements are gaining traction, as they leverage the strengths of both empirical data and established biological knowledge [11].
As multi-omics technologies continue to evolve, particularly in single-cell and spatial omics, network integration methods must adapt to handle increasing data complexity and resolution. Future developments will likely focus on dynamic network models that can capture temporal changes in biological systems, as well as multi-scale approaches that can integrate data from molecular, cellular, and tissue levels [12]. These advances will further enhance our ability to map the complex relationships between biological layers and translate these insights into improved diagnostic and therapeutic strategies.
Diagram 2: Network integration process mapping multi-omics data to biological insights
Frequently Asked Questions (FAQs)
Q1: What is the first thing I should check if my multi-omics data integration fails? Your first step should be to verify data harmonization. Ensure all datasets have been standardized and preprocessed, which includes normalization, batch effect correction, and conversion to compatible formats and units. Incompatible data formats or scales are a leading cause of integration failure [15].
Q2: I'm getting a "module not found" error for OmicsIntegrator. How can I resolve this?
This error is typically environment-related. Confirm you are using a Linux OS, as this is the primary supported development environment. Provide your sessionInfo() or package version details when seeking help, as this is required for others to reproduce your issue [45].
Q3: Our federated analysis is producing inconsistent results across sites. What could be the cause? Inconsistent results in federated analytics often stem from a lack of harmonized data standards and governance across participants. Implement shared protocols for data formats, quality control, and processing workflows. Effective federation requires central teams to provide shared infrastructure and governance to ensure consistency, while embedded teams handle local analysis [46].
Q4: Why is my multi-omics resource difficult for other researchers to use? This common pitfall occurs when resources are designed from the data curator's perspective rather than the end-user's. To avoid this, design your resource around real user scenarios from the beginning. Pretend you are an analyst trying to solve a specific biomedical problem and structure your resource to meet those needs [15].
Q5: What are the key differences between federated analysis, federated learning, and federated analytics? These are distinct but related approaches:
- Federated Data Analysis: The comprehensive umbrella term for running analyses on distributed data, sharing only results [47].
- Federated Learning: Focuses specifically on training machine learning models across distributed data by sharing model updates (e.g., gradients), not raw data [47].
- Federated Analytics: Deals with performing basic statistical computations (e.g., averages, counts) on distributed datasets [47].
Troubleshooting Guides
Common Installation and Environment Issues
Problem: OmicsIntegrator web version is unavailable.
- Solution: The web version has been taken down for maintenance. Use the command-line versions available on GitHub instead [48].
Problem: R package errors in Windows OS.
- Solution: Switch to a Linux environment. Many bioinformatics packages, including those for multi-omics analysis, are developed and tested primarily on Linux. Some OS-specific low-level issues are difficult to debug outside this environment [45].
Data Processing and Integration Errors
Problem: Failure to integrate unmatched multi-omics data (from different cells).
- Solution: Unmatched data requires specialized diagonal integration tools that project cells into a co-embedded space. Use tools like GLUE, Pamona, or Seurat v5's Bridge Integration designed for this purpose [30].
Problem: Integrated data resource is underutilized by the scientific community.
- Solution: Redesign your resource from the user's perspective, not the curator's. Create real use case scenarios and ensure your resource effectively solves these problems. Document every step and make software code openly available [15].
Federated Analysis Challenges
Problem: Difficulty establishing a federated analytics operating model.
- Solution: Implement a structured model with three layers [46]:
- A central team for shared infrastructure and governance
- Embedded teams within business units for local insights
- Governance and community mechanisms for alignment and trust
Essential Research Reagent Solutions
Table: Key Computational Tools for Multi-Omics Integration and Federated Analysis
| Tool Name | Primary Function | Key Features | Use Case |
|---|---|---|---|
| OmicsIntegrator [48] [6] | Network-based data integration | Prize-Collecting Steiner Forest algorithm to identify high-confidence subnetworks | Identifying cellular pathways and relevant proteins from proteomic data |
| MOFA+ [30] [15] | Factor analysis | Unsupervised integration of multiple omics layers; handles missing data | Vertical integration of matched multi-omics data from the same samples |
| GLUE [30] | Graph-linked unified embedding | Uses prior biological knowledge to anchor features; enables triple-omic integration | Unmatched (diagonal) integration of different omics from different cells |
| Seurat v4/v5 [30] | Weighted nearest neighbor & bridge integration | Integrates mRNA, spatial coordinates, protein, accessible chromatin | Both matched and unmatched integration scenarios |
| DataSHIELD [47] | Privacy-preserving federated analysis | R-based with built-in privacy protections; no cryptography expertise needed | Federated analysis of sensitive data across multiple institutions |
| mixOmics [15] | Multivariate data integration | R package for large-scale omics data integration; multiple statistical methods | Horizontal integration of the same omic type across multiple datasets |
Multi-Omics Data Integration Workflow
The following diagram illustrates a robust workflow for multi-omics data integration, emphasizing best practices for data harmonization.
Best Practices for Data Harmonization
Standardization and Preprocessing Protocol
Data Normalization: Account for differences in sample size, concentration, and measurement units across platforms [15].
Batch Effect Correction: Remove technical biases or artifacts introduced by different experimental batches or platforms [15].
Quality Control Filtering: Remove outliers or low-quality data points while documenting all filtering criteria [15].
Metadata Annotation: Provide comprehensive metadata describing samples, equipment, and software used, as metadata facilitates data search and retrieval [15].
Format Unification: Convert diverse data formats to a unified samples-by-feature matrix (n-by-k) compatible with machine learning and statistical methods [15].
Federated Data Management Protocol
Central-Embedded Model: Establish clear responsibilities where central teams manage shared infrastructure and governance while embedded teams deliver business-specific insights [46].
Data Harmonization: Before federated analysis begins, ensure all participants agree on data formats, standards, and ontologies [47].
Privacy-Preserving Technologies: Implement appropriate safeguards such as differential privacy, secure multiparty computation, or homomorphic encryption based on data sensitivity [47].
MVP Handoff Mechanism: Create pathways for local minimum viable products (MVPs) to be evaluated for broader use, then hardened and maintained by central teams [46].
Navigating Pitfalls: Solving Common Data Quality and Technical Hurdles
Troubleshooting Guides
Troubleshooting Guide for Incomplete Data
User Question: "A significant portion of patient demographic data in our integrated multi-omics dataset is missing. How can we identify the root cause and remedy this?"
| Problem Identification | Root Cause Analysis | Remediation Protocol | Validation & Quality Control |
|---|---|---|---|
| Quantify Missingness: Profile data to calculate the percentage of empty values for each key variable (e.g., age, gender) [49]. | Review Data Entry: Check if missingness is random or systematic (e.g., all missing from one source site) [50]. | Preventive Controls: Implement required fields in electronic data capture (EDC) systems to block record submission until key fields are complete [50]. | Automated Monitoring: Use tools to continuously track the "number of empty values" metric, alerting when thresholds are breached [51] [52]. |
| Assess Impact: Determine if incomplete records bias downstream analyses or cohort building [53]. | Audit Source Systems: Identify if the issue stems from system incompatibilities during data integration [49]. | Data Augmentation: Attempt to complete missing fields by comparing with a known accurate dataset [50]. | Curation Review: For shared data, have data curators assess completeness as part of repository quality assurance [53]. |
Troubleshooting Guide for Inaccurate Data
User Question: "We suspect inaccuracies in transcriptomic sample identifiers, leading to incorrect sample-to-patient mappings. What is the best protocol to address this?"
| Problem Identification | Root Cause Analysis | Remediation Protocol | Validation & Quality Control |
|---|---|---|---|
| Validate Against Source: Cross-check a subset of sample IDs against original laboratory records or pre-COVID-19 cohort data for discrepancies [10] [53]. | Trace Data Lineage: Use lineage tools to track the data's journey and pinpoint the transformation or transfer step where inaccuracies were introduced [52]. | Automate Data Entry: Minimize human error by automating data transfer from source instruments to analysis databases where possible [50]. | Implement Data Quality Tools: Deploy solutions like Great Expectations or Soda Core to run automated validation checks (e.g., checking ID format conformity) against predefined rules [51] [52]. |
| Calculate Error Ratio: Compute the "data to errors ratio" to understand the scale of inaccuracy relative to the dataset size [51]. | Check for Stale Data: Assess if data has decayed over time, a common cause of inaccuracy [50] [49]. | Isolate or Delete: Use a tool like DataBuck to identify and quarantine inaccurate data. If it cannot be fixed by comparing with a trusted source, delete it to prevent contamination of analysis [50]. | FAIR Principles: Ensure corrected data is supported by rich metadata to promote appropriate interpretation and reuse, a key aspect of data quality [10] [53]. |
Troubleshooting Guide for Inconsistent Data
User Question: "After merging genomic and proteomic datasets from different platforms, we have inconsistent formatting for genetic variants and date fields. How do we resolve this?"
| Problem Identification | Root Cause Analysis | Remediation Protocol | Validation & Quality Control |
|---|---|---|---|
| Profile Data Formats: Use data profiling tools to identify inconsistencies in dates (e.g., MM/DD/YYYY vs. DD-MON-YY), units of measurement, and nomenclature [51] [49]. | Audit Source Systems: Identify cross-system inconsistencies by reviewing the data formats and standards used by each originating omics platform [50] [6]. | Adopt Common Data Elements (CDEs): Define and implement standardized concepts that precisely define variables with a specified set of responses across all studies [10]. | Programmatic Validation: Use R or Python scripts to validate data structure, format, adherence to controlled terminologies, and conditional field consistency post-harmonization [10]. |
| Check Logical Consistency: Look for conflicts, such as a sample date recorded before a patient's birth date [53]. | Map Harmonization Challenges: Document where study-specific variables have no corresponding CDE, leading to uneven adoption [10]. | Retrospective Harmonization: Programmatically transform raw study data to align with the CDEs and a single, standardized format [10] [50]. | Quality Control Evaluation: Upload harmonized data to a cloud-based ecosystem like BioData Catalyst for quality control and peer review [10]. |
Experimental Protocols for Data Harmonization
Detailed Protocol: Retrospective Harmonization to Common Data Elements
This protocol is derived from the experiences of the NHLBI CONNECTS program, which harmonized COVID-19 clinical trial data for sharing on the BioData Catalyst ecosystem [10].
1. Pre-Harmonization Assessment
- Define Scope: Establish clear research objectives and define the omics datasets to be integrated [6].
- Create Harmonization Template: Develop a mapping template in collaboration between data managers and statisticians. This template guides the transformation of source variables to target CDEs.
- Secure Resources: This process is labor-intensive and can delay data sharing by several months; secure appropriate staff and computational resources [10].
2. Variable Mapping and Transformation
- Programmatic Mapping: Most studies implement harmonization instructions programmatically using languages like SAS or R. The raw data is transformed according to the harmonization template.
- Address Subjectivity: Mitigate mapping bias by involving diverse perspectives (e.g., domain experts, bioinformaticians) to establish content equivalence across different study designs and collection instruments [10].
- Handle Incompatible Data: Document instances where mapping is not possible due to incompatible scales or study-specific variables with no corresponding CDE.
3. Validation and Quality Control
- Automated Validation: Execute an R script (or equivalent) to programmatically evaluate each CDE domain. The checks should assess:
- Data Structure and Format: Data type, length.
- Required Columns: Presence of all mandatory fields.
- Controlled Responses: Adherence to predefined value sets.
- Data Integrity: Missingness patterns and conditional field consistency.
- Assign Validation Status: Assign a "Pass," "Fail," or "Warning" status to each field based on the validation checks. A "Warning" may indicate excessive missingness or values outside an expected range that require human review [10].
4. Data Packaging and Sharing
- Export Data: Export harmonized data into widely accessible, non-proprietary formats (e.g., comma-delimited files).
- Provide Comprehensive Metadata: Support the dataset with rich metadata, including key indices for effective search and context for appropriate interpretation [10].
- Share Raw and Harmonized Data: For maximal transparency and interoperability, publicly share both the raw (as-collected) and harmonized (CDE-mapped) datasets [10].
Workflow Visualization
Data Quality Framework
The Scientist's Toolkit: Research Reagent Solutions
| Tool or Software | Category | Primary Function | Relevance to Multi-Omics Data Harmonization |
|---|---|---|---|
| Great Expectations [51] [52] | Open-Source Data Validation | Creates "unit tests for data"; defines and validates expectations for data quality (e.g., null checks, value ranges). | Testing and documenting data pipelines to ensure ingested omics data meets predefined quality standards before integration. |
| Soda Core [51] [52] | Open-Source Data Quality | Uses a simple YAML syntax (SodaCL) to define data quality checks and scan datasets for issues. | Accessible quality checks for data analysts and scientists to profile individual omics datasets and identify formatting flaws. |
| dbt Core [51] | Open-Source Transformation | Performs built-in data quality tests within data transformation pipelines in a data warehouse. | Embedding quality checks (e.g., uniqueness, acuracy) directly into the SQL-based transformation workflows that prepare omics data for analysis. |
| Monte Carlo [51] [52] | Data Observability Platform | Uses machine learning to automatically detect data anomalies across the entire pipeline (freshness, volume, schema). | Providing end-to-end visibility into the health of multi-omics data pipelines, catching issues like broken data streams before they impact analyses. |
| Common Data Elements (CDEs) [10] | Standardization Framework | Standardized concepts that precisely define questions and specified responses. | The foundational element for harmonizing variables across different clinical trials and omics studies to ensure interoperability. |
| OmicsIntegrator [6] | Multi-Omics Analysis Tool | Provides robust data integration capabilities for harmonizing diverse multi-omics datasets. | Streamlining the technical process of combining genomic, transcriptomic, proteomic, and metabolomic data into a unified dataset. |
Frequently Asked Questions (FAQs)
What are the most common data quality issues in integrated datasets? The most frequent issues are inaccurate data (wrong or erroneous entries), incomplete data (missing values in key fields), and inconsistent data (formatting or unit mismatches across sources) [50] [49]. Other common problems include duplicate records, outdated (stale) data, and unstructured data that doesn't conform to a standard schema [50] [49].
How can we proactively prevent data quality issues during study design? The most effective strategy is up-front standardization. Adopt Common Data Elements (CDEs) during the study design phase to ensure all data is collected consistently from the start [10]. Implementing required fields in electronic data capture systems and automating data entry from instruments also significantly reduces future errors [50].
What is the difference between data standardization and data harmonization? Standardization is the process of defining and implementing common data formats, protocols, and elements before data is collected. Harmonization is the retrospective process of aligning and transforming data that was collected using different standards into a common format for integrated analysis [10]. Harmonization is often more complex and resource-intensive.
Why is it important to share both raw and harmonized datasets? Sharing both datasets maximizes transparency and interoperability. The raw data represents the data as originally collected, preserving its original state. The harmonized data provides a version that is consistent and comparable with other studies, enabling immediate reuse and collaborative analysis [10]. This practice allows other researchers to understand the transformations applied and gives them the flexibility to use the data as needed.
What metrics should we track to monitor data quality over time? Key data quality metrics to track include [51]:
- Number of Empty Values: The count of missing fields in a dataset.
- Data to Errors Ratio: The number of known errors relative to the dataset size.
- Schema Conformance: The percentage of data that conforms to the expected format. Consistently tracking these metrics helps quantify data health and identify degrading quality before it impacts research outcomes.
Overcoming the High-Dimensionality, Low-Sample-Size (HDLSS) Challenge
FAQs on the HDLSS Problem in Multi-Omics
What is the HDLSS problem, and why is it so common in multi-omics research? The HDLSS problem occurs when the number of features (dimensions) in a dataset is vastly greater than the number of samples. In multi-omics, a single -omic dataset can contain tens of thousands of features (e.g., over 20,000 human genes from RNAseq), while most studies contain only a few hundred samples [54]. This imbalance violates the ideal condition for machine learning (ML), which performs better with more samples than features [54].
How does data harmonization help mitigate the HDLSS challenge? Data harmonization reconciles different datasets by standardizing their syntax (data formats), structure (conceptual schema), and semantics (intended meaning) [7]. This process is crucial before data integration. For HDLSS, proper harmonization includes dimensionality reduction and normalization, which help reduce noise and the overall feature count, making the data more tractable for ML models [54] [15].
What are the most common machine learning techniques used for HDLSS data? Popular ML techniques identified in the literature are those suited to datasets with many features and few samples. These include autoencoders (a type of neural network for dimensionality reduction), random forests, and support vector machines [54].
Troubleshooting Common HDLSS Scenarios
Problem: My multi-omics model is overfitting.
- Potential Cause: The high number of features relative to samples allows the model to memorize noise instead of learning generalizable patterns.
- Solution: Apply dimensionality reduction techniques as a preprocessing step. Methods like autoencoders are explicitly used in multi-omics to compress the data into a lower-dimensional space, effectively reducing the number of features [54].
Problem: Integrating my omics datasets creates a huge, unmanageable matrix.
- Potential Cause: You are likely using an "Early Integration" strategy, which simply concatenates all datasets into one large matrix, drastically increasing dimensionality without adding new samples [8].
- Solution: Consider an "Intermediate Integration" strategy. This approach integrates datasets to find a common representation while accounting for data-specific factors, which can reduce noise and heterogeneity [8].
Problem: My data comes from different platforms and has inconsistent formats.
- Potential Cause: A lack of standardization and harmonization in the initial data processing phase.
- Solution: Preprocess your data to ensure it is standardized and harmonized [15]. This involves:
- Normalization: Account for differences in sample size, concentration, and technical biases [15].
- Batch Effect Correction: Remove technical variations that are not due to biological factors [15].
- Mapping to Ontologies: Use domain-specific ontologies to align the semantic meaning of features across datasets [15] [7].
Multi-Omics Data Landscape
The table below summarizes the prevalence of different omics data types and the typical scale of features and samples involved, highlighting the source of the HDLSS challenge [54].
| Omics Data Type | Prevalence in Studies | Typical Number of Features | Typical Number of Samples |
|---|---|---|---|
| Transcriptomics | 42% (Most popular) | Tens of thousands (e.g., >20,000 genes) | A few hundred (Median: 447) |
| Epigenomics | 22% | Often very high | A few hundred |
| Genomics | 21% | Often very high | A few hundred |
| Proteomics | 6% | Hundreds to thousands | A few hundred |
| Metabolomics | 2% | Hundreds to thousands | A few hundred |
Experimental Protocols for HDLSS Data
Protocol 1: Dimensionality Reduction using an Autoencoder Autoencoders are a popular deep learning method for compressing high-dimensional omics data [54].
- Input: Your high-dimensional omics data matrix (samples-by-features).
- Encoding: The data is fed through an "encoder" network that compresses it into a lower-dimensional "bottleneck" layer (the latent space).
- Decoding: A "decoder" network attempts to reconstruct the original input from this compressed representation.
- Training: The model is trained by minimizing the difference between the original input and the reconstruction.
- Output: After training, the encoder can be used to transform your high-dimensional data into the lower-dimensional latent space, which becomes the new input for your classifier or regression model.
Protocol 2: Data Harmonization for Multi-Omics Integration This protocol ensures data from different omics platforms are comparable [15] [7].
- Standardization:
- Harmonization:
- Batch Effect Correction: Use statistical methods (e.g., ComBat) to remove non-biological variations introduced by different processing batches [15].
- Semantic Alignment: Map gene IDs, protein names, and other identifiers to a common ontology (e.g., KEGG, RefSeq) to ensure features are comparable across datasets [15] [42].
- Validation: Use exploratory data analysis (e.g., PCA plots) before and after harmonization to confirm the reduction of technical artifacts.
Data Harmonization Workflow
The following diagram illustrates the logical workflow for preparing multi-omics data to overcome the HDLSS challenge.
The Scientist's Toolkit
The table below lists key computational and methodological "reagents" essential for tackling the HDLSS problem.
| Tool / Method | Function | Application Context |
|---|---|---|
| Autoencoders | A neural network for non-linear dimensionality reduction. | Compressing high-dimensional omics data (e.g., transcriptomics) into a lower-dimensional latent representation before classification [54]. |
| Random Forests | An ensemble ML method robust to noise and overfitting. | Building classifiers or regressors directly on HDLSS data; can provide feature importance scores [54]. |
| mixOmics (R) | A toolkit for the exploration and integration of omics data. | Performing multivariate dimensionality reduction and integration for multi-omics datasets [15]. |
| INTEGRATE (Python) | A Python tool for multi-omics data integration. | Implementing various data integration strategies in a Python workflow [15]. |
| Variational Autoencoders | A probabilistic method for data harmonization. | Aligning datasets from different batches or platforms by learning a shared latent structure [15]. |
| MultiPower | An open-source tool for sample size estimation. | Calculating the statistical power and optimal sample size for a planned multi-omics study [42]. |
Addressing Data Heterogeneity and Siloed System Integration
Frequently Asked Questions
What are the most common sources of data heterogeneity in multi-omics studies? Data heterogeneity arises from differences in syntax (file formats like .csv, JSON), structure (data organized as event data vs. panel data), and semantics (differing definitions for the same term across datasets) [7]. Technically, variations arise from different omics platforms, measurement units, sample collection methods, and sample processing protocols, leading to batch effects and distribution shifts that impede direct data combination [55] [56] [15].
How can I quickly assess if my datasets are suffering from significant batch effects? Initial assessment can involve unsupervised methods like Principal Component Analysis (PCA). If samples cluster strongly by batch (e.g., date of processing, sequencing run) rather than by biological condition, this indicates significant batch effects. For a more quantitative approach, use discrepancy measurement techniques like Maximum Mean Discrepancy (MMD) to quantify the distributional difference between batches before and after applying correction methods [55].
We have data from different omics platforms. Should we use data-driven or model-driven integration methods? The choice depends on your data characteristics and research goals. The table below compares the two approaches [55].
| Feature | Data-Driven Methods | Model-Driven Methods |
|---|---|---|
| Best For | Homogeneous, well-represented datasets; baseline modeling [55] | Heterogeneous datasets; capturing complex interdependencies [55] |
| Common Techniques | Direct concatenation, matrix factorization, CCA [55] | Deep neural networks, probabilistic fusion, domain adaptation [55] |
| Advantages | Simplicity, scalability, practicality with limited domain priors [55] | Interpretability, ability to learn shared feature representations [55] |
| Disadvantages | Risk of overfitting, difficulty with heterogeneous data [55] | Requires more information (e.g., dataset interactions) [55] |
What is the fundamental difference between data harmonization and data integration? Data harmonization reconciles conceptually similar datasets into a single, cohesive ontology (e.g., combining multiple COVID-19 policy datasets into one). Data integration or linkage combines conceptually different datasets into a multidimensional resource (e.g., merging COVID-19 data, economic data, and clinical outcomes) [7].
Troubleshooting Guides
Problem: Batch Effects and Technical Variation
Issue: Your data shows strong technical artifacts from different processing batches that obscure biological signals.
Solution:
- Diagnose: Use PCA to visualize batch-related clustering.
- Apply Batch Correction:
- For genomic data, use established tools like ComBat or Limma, which estimate and adjust for batch effect parameters like mean and variance [55]. Note that these methods often assume identical cell population compositions across batches, which may not hold true [55].
- For more complex distribution shifts, employ Domain Adaptation techniques like Domain-Adversarial Neural Networks (DANN). These methods learn batch-invariant feature representations by minimizing the discrepancy between source and target domains [55].
- Validate: Ensure that after correction, biological groups separate better in visualizations, and known biological truths are preserved.
Problem: Semantic Inconsistencies Across Datasets
Issue: The same term (e.g., "young adult") has different definitions across datasets, or different terms describe the same concept.
Solution:
- Create a Data Dictionary: Before integration, define a unified ontology or taxonomy for all key concepts in your study [7] [57].
- Map Terminology: Systematically map all variable names and definitions from the source datasets to this common dictionary. This can be a stringent process (using identical measures) or a flexible one (ensuring inferential equivalence) [7].
- Leverage Standardized Ontologies: Use community-accepted ontologies (e.g., Gene Ontology) to ensure semantic interoperability [15] [57].
Problem: Integrating Data with Different Structures and Formats
Issue: Data is locked in siloed systems with incompatible formats (e.g., event data vs. panel data, .csv vs. JSON).
Solution:
- Standardize and Preprocess: Convert all data into a compatible format. This involves:
- Apply Data Harmonization: Resolve structural heterogeneity. For example, transform panel data (where a single event spans multiple rows) into an event-data format (one row per event) or vice versa to ensure compatibility [7].
- Store Raw Data: Always keep the raw data accessible to ensure full reproducibility of the preprocessing and harmonization steps [15].
The following workflow diagram outlines the core process for addressing data heterogeneity.
Problem: Missing Data in Multi-Omics Datasets
Issue: Missing values for some omics layers in a subset of samples, creating an incomplete picture.
Solution:
- Experimental Design: The best strategy is prevention. Plan multi-omics studies to generate data from the same set of samples where possible, ensuring sufficient biomass for all planned assays [56].
- Imputation and Advanced Methods: When data is missing, consider:
- Model-Driven Imputation: Use methods like coupled matrix and tensor factorization to transfer information learned from one complete dataset to impute missing values in another [55].
- Collective Matrix Factorization (CMF): This technique simultaneously factorizes several matrices, allowing datasets to share the same latent space and efficiently handle missing data [55].
The Scientist's Toolkit: Key Research Reagents & Solutions
The following table details essential computational tools and methods for tackling data integration challenges.
| Tool/Method Name | Function | Use Case |
|---|---|---|
| ComBat [55] | Removes batch effects by estimating and adjusting for batch-specific parameters. | Correcting for technical variation in genomic and transcriptomic data. |
| Domain-Adversarial Neural Networks (DANN) [55] | A domain adaptation method that learns features indistinguishable between source and target domains. | Adapting models trained on one dataset (source) to perform well on another with different distributions (target). |
| Coupled Matrix/Tensor Factorization [55] | Jointly factorizes multiple data matrices to share information and impute missing values. | Integrating partially coupled data from multiple platforms (e.g., genomics and proteomics). |
| mixOmics (R) / INTEGRATE (Python) [15] | Provides a framework for multivariate analysis and integration of multiple omics datasets. | Exploratory data analysis and supervised integration of diverse omics data types. |
| Conditional Variational Autoencoders (cVAE) [15] | A deep learning approach for data harmonization using style transfer. | Harmonizing data from different sources, such as RNA-seq data from different labs. |
| Logic Forest [58] | A machine learning algorithm to identify salient main effects and interactions between factors. | Discovering interactions between genetic and environmental risk factors in disease outcomes. |
Best Practices for Scalable Data Pipelines and Maintenance
FAQs on Data Pipelines in Multi-Omics Research
Q: What is a data pipeline in the context of multi-omics research? A: A data pipeline is a series of steps that moves data from source systems to a destination for storage and analysis. In multi-omics, this involves ingesting, transforming, and integrating disparate data types (genomics, transcriptomics, proteomics, etc.) into a cohesive, analysis-ready dataset. This process is critical for creating a unified view of biological systems [59].
Q: Why is a modular pipeline design important for multi-omics studies? A: Modular design, where a pipeline is broken into independent, reusable components (e.g., separate ingestion, transformation, and quality control modules), makes pipelines easier to test, update, and maintain. This is essential in multi-omics due to the variety of data types and rapid evolution of analytical technologies, allowing researchers to adapt workflows without rebuilding them entirely [60].
Q: How can we ensure data quality in high-throughput omics pipelines? A: Implement automated data quality checks and validation at every stage of the pipeline. This includes profiling raw data upon ingestion, validating transformations, and using open-source libraries to run checks for completeness, accuracy, and consistency. Preventing poor-quality data from propagating is vital to avoid distorted biological insights [61] [60].
Q: What is the role of a "dead-letter queue" in a data pipeline? A: A dead-letter queue is a pattern for robust error handling. Instead of failing or dropping data that causes processing errors (e.g., due to unexpected schemas or formatting), the problematic data is routed to a separate, monitored destination. This preserves the data for later inspection and troubleshooting, ensuring the main pipeline continues to run and data is not lost [62].
Q: What are the biggest challenges in building scalable multi-omics data pipelines? A: Key challenges include integrating disparate and heterogeneous data sources, ensuring data harmonization across different omics layers, and managing the immense volume and complexity of data. Furthermore, a lack of observability can make it difficult to detect anomalies or trace root causes, eroding trust in the data's reliability [59] [5].
Troubleshooting Common Data Pipeline Issues
The table below summarizes frequent data pipeline failures, their root causes, and recommended solutions, synthesized from studies of data pipeline projects [60].
| Issue | Frequency | Root Cause | Solution |
|---|---|---|---|
| Data Type Errors | 33% of projects | Data arrives in a format different from what is expected (e.g., text in a numeric field). | Implement schema validation and automated data profiling at ingestion; use data quality tools. |
| Misplaced Characters | 17% of projects | Stray symbols (e.g., extra commas, quotes) break the data structure during parsing. | Use parallel parsers that can detect and quarantine syntax errors without stopping the entire pipeline. |
| Raw Data Issues | 15% of projects | Missing values, data duplication, or corrupted data during ingestion. | Introduce data quality checks for completeness and uniqueness; establish data contracts with data providers. |
| Integration Challenges | 29% of projects | Difficulties transforming data across databases and aligning different platforms or languages. | Adopt a modular pipeline design and use standardized data models to simplify integration tasks. |
| Ingestion & Loading Issues | 18% of projects each | Problems connecting to source databases; slow or incorrect data loading. | Use optimized data connectors and efficient, columnar data formats (e.g., Parquet) for storage. |
Experimental Protocol: Implementing a Data Quality Framework
This protocol provides a detailed methodology for establishing a robust data quality framework within a multi-omics data pipeline.
1. Objective To systematically validate data across key dimensions—completeness, accuracy, validity, and consistency—at each stage of the multi-omics data pipeline to ensure the integrity of downstream analyses.
2. Materials and Reagents
- Computing Infrastructure: Access to a high-performance computing (HPC) cluster or cloud computing platform (e.g., AWS, GCP).
- Data Quality Tool: Installation of an open-source data quality library such as Amazon Deequ or Great Expectations.
- Source Data: Raw multi-omics datasets (e.g., FASTQ, BAM, or mass spectrometry files).
3. Methodology
- Step 1: Define Quality Metrics. Before pipeline execution, define specific quality checks for each data type. For example:
- Completeness: Check for missing values in required fields (e.g., sample ID, gene expression value).
- Accuracy/Validity: Validate that data falls within expected ranges (e.g., DNA methylation values between 0 and 1).
- Consistency: Verify that gene identifiers are consistent across different omics datasets.
- Uniqueness: Ensure no duplicate records for primary keys.
- Step 2: Integrate Checks into Pipeline. Incorporate the defined checks as automated steps within the pipeline workflow using your chosen tool. For instance, with Deequ, you can declare checks that run on each new data partition.
- Step 3: Configure Error Handling. Implement a dead-letter queue pattern. When a data unit fails a quality check, the pipeline should log the error and route the failed data to a designated storage location (e.g., a specific database table or cloud storage bucket) for further investigation, while allowing valid data to proceed.
- Step 4: Continuous Monitoring. Set up automated monitoring and alerting (e.g., using Grafana dashboards) to track data quality metrics over time. Configure alerts to notify engineers or researchers when metrics drift beyond acceptable thresholds [61] [60].
Data Harmonization Workflow for Multi-Omics
The diagram below illustrates the logical flow for harmonizing disparate multi-omics data into an integrated, analysis-ready resource.
The Scientist's Toolkit: Essential Research Reagents & Solutions
This table details key computational tools and resources essential for building and maintaining robust multi-omics data pipelines.
| Tool / Resource | Function | Application in Multi-Omics |
|---|---|---|
| dbt (Data Build Tool) | A transformation tool that uses SQL to build modular, tested, and documented data models inside the data warehouse. | Enables clean, version-controlled transformation of raw omics data into analyzable models, facilitating ELT (Extract, Load, Transform) workflows [59]. |
| Apache Airflow / Dagster | Orchestration platforms used to schedule, manage, and monitor complex data workflows as directed acyclic graphs (DAGs). | Coordinates the execution of multiple, dependent data processing steps across different omics data types, ensuring workflows run in the correct order and time [59] [60]. |
| Amazon Deequ / Great Expectations | Open-source libraries for defining and automating data quality checks based on metrics like completeness and uniqueness. | Implements "unit tests" for large-scale omics datasets, validating data upon arrival and blocking jobs if quality thresholds are not met [60]. |
| Datahub / Atlan | Metadata management and data discovery platforms that provide data lineage, governance, and search capabilities. | Offers visibility into the origin and transformation journey of omics data, building trust and helping researchers discover and understand available datasets [60]. |
| Parquet File Format | An efficient, open-source columnar storage format optimized for analytical querying and large-scale data processing. | Reduces storage costs and improves I/O performance when storing and querying massive omics datasets (e.g., from whole genome sequencing) [60]. |
Ensuring Data Security, Privacy, and Ethical Governance in Federated Analyses
Frequently Asked Questions (FAQs)
General Principles
Q1: What is federated analysis, and how does it fundamentally enhance data privacy?
Federated analysis is a computational paradigm where the analysis (via algorithms or models) is brought to the data, rather than moving sensitive data to a central repository. In this model, queries and computation code are sent to distributed data sources for local execution. Only aggregated, non-identifiable results are returned to the researcher [63]. This fundamentally enhances privacy by ensuring that raw, individual-level data never leaves the secure control of the data owner, significantly reducing the risk of data breaches and re-identification [64] [63].
Q2: Beyond technology, what are the core pillars of governance for a federated project?
Effective governance for a federated project rests on three core pillars [65] [66]:
- Procedural Mechanisms: The policies and procedures that guide the project, including data privacy measures (e.g., de-identification standards), formal data use agreements, and ongoing monitoring and auditing.
- Relational Mechanisms: The frameworks for collaboration among all stakeholders (data owners, researchers, platform operators) to ensure clear communication and trust.
- Structural Mechanisms: The defined roles, responsibilities, and technical infrastructure. This includes establishing a governance council, clear data ownership, and implementing interoperable data standards like the OMOP common data model or HL7 FHIR [65].
Technical Implementation & Troubleshooting
Q3: We are observing a significant drop in our federated model's accuracy. Could our privacy-preserving techniques be the cause?
Yes, this is a known challenge in the privacy-utility trade-off. If you are using Differential Privacy (DP), the calibrated noise added to the gradients or model updates to protect privacy can degrade model utility [64] [67]. To troubleshoot:
- Audit your Privacy Budget (
ε): A very lowε(strong privacy) requires more noise. Re-evaluate if your privacy budget is too stringent for your accuracy requirements [67]. - Consider a Hybrid Approach: For clients with sufficient computational resources, using Homomorphic Encryption (HE) can contribute noise-free updates, potentially improving overall model accuracy without sacrificing privacy. A proposed PPML-Hybrid method allows this flexibility [67].
- Explore Techniques like Shuffling: Methods like PPML-Omics use Decentralized Randomization (shuffling updates among clients) which can amplify the privacy guarantee, allowing you to achieve the same level of protection with less noise [64].
Q4: What are the primary privacy attacks against federated learning, and how can we defend against them?
Federated models are vulnerable to several novel attacks [64] [65]. The table below summarizes common attacks and defense strategies.
Table: Privacy Attacks and Defense Mechanisms in Federated Learning
| Attack Type | Description | Defense Strategies |
|---|---|---|
| Membership Inference [64] | An attacker determines whether a specific individual's data was part of the training set. | Implement Differential Privacy (DP) to obfuscate the influence of any single data point [64] [67]. |
| Model Inversion / Data Reconstruction [64] | An attacker reverse-engineers the model's updates to reconstruct sensitive raw training data. | Use Homomorphic Encryption (HE) to aggregate encrypted gradients, preventing a "curious" server from seeing individual updates [67]. |
| Model Poisoning [65] | A malicious participant submits corrupted model updates to degrade the global model's performance or introduce biases. | Implement robust aggregation algorithms and continuous monitoring to detect and filter out anomalous updates [65]. |
Q5: How can we handle the high computational cost of privacy technologies like Homomorphic Encryption?
The computational overhead of HE is a significant practical constraint [67]. To mitigate this:
- Adopt a Hybrid System: Allow resource-constrained clients to use the less computationally intensive DP method, while only well-resourced clients use HE. This provides a balance between efficiency and the accuracy benefits of noise-free updates [67].
- Optimize Cryptographic Operations: Leverage efficient HE schemes like CKKS (Cheon-Kim-Kim-Song), which is designed for approximate arithmetic on real numbers, making it suitable for machine learning tasks [67].
- Strategic Use: Reserve HE for the most sensitive data or critical model parameters rather than applying it to the entire dataset or model.
Governance, Compliance, and Ethics
Q6: How can we ensure our federated analysis complies with evolving global data regulations?
Compliance requires a proactive, multi-layered approach:
- Privacy by Design: Integrate privacy protections into the system architecture from the start, not as an afterthought. This includes principles like data minimization and storage limitation, where you collect only the necessary data and retain it only for as long as needed [68].
- Stay Informed on Regulations: Be aware that 2025 sees continued strict enforcement and new laws, such as more U.S. state privacy laws offering special protections for teen data, and the full implementation of the EU AI Act [69] [68].
- Use Binding Legal Agreements: Establish robust Data Use Agreements (DUAs) between all participating organizations that clearly define the scope of permitted research, data security requirements, and liability [63] [65].
Q7: What are the best practices for managing data access in a multi-institutional federation?
A successful access model combines technology and governance [63] [65]:
- Centralized Data Catalog: Provide a searchable metadata repository so researchers can discover available datasets without seeing the raw data.
- Formal Access Request Workflow: Researchers must submit a proposal with their research question and analysis plan.
- Approval by Data Access Committees (DAC): Each data-owning institution should have a DAC that reviews requests against ethical and institutional policies before granting access.
- Role-Based Access Control: Technical systems should enforce permissions, ensuring researchers can only run analyses on data for which they have explicit approval.
Troubleshooting Guides
Issue 1: Data Heterogeneity and Harmonization Errors
Problem: Models trained across different sites show poor performance and low generalizability due to inconsistent data formats, coding standards, and pre-processing pipelines.
Solution:
- Implement a Common Data Model (CDM): Mandate the use of a standard model like the OMOP CDM to ensure semantic consistency across all data partners. This ensures that a term like "myocardial infarction" is interpreted uniformly [65].
- Develop Harmonization Pipelines: Create and share automated workflows that transform local raw data into the standardized CDM format. These pipelines should be run by each data owner before their data is included in the federation [63].
- Use Shared Ontologies: Adopt standardized vocabularies (e.g., SNOMED CT for clinical terms) to further enhance interoperability [63].
The following workflow diagram illustrates a robust data harmonization and federated analysis process:
Issue 2: Establishing Trust in a Decentralized Network
Problem: Data owners are hesitant to participate due to concerns about how their data will be used and protected by other parties in the federation.
Solution:
- Choose a Trust Model: Decide on a governance structure. A centralized trust model with a highly trusted coordinator (e.g., a national health institute) can vet participants. Alternatively, a federated governance model combines central oversight with domain-level ownership and control [63] [66].
- Ensure Transparency and Auditing: Maintain immutable logs of all queries and data access events. Allow for regular third-party audits to verify that all activities comply with the agreed-upon policies [65].
- Create a Clear Code of Conduct: Develop a binding agreement for all participants that outlines acceptable use, security protocols, and incident response plans [63].
Issue 3: Implementing a Balanced Privacy-Preserving Strategy
Problem: Choosing between Differential Privacy (DP) and Homomorphic Encryption (HE) involves a difficult trade-off between privacy strength, model utility, and computational cost.
Solution: Implement a hybrid strategy that allows for client flexibility. The following diagram outlines the decision process for the PPML-Hybrid method, which balances these factors [67].
Table: Comparison of Privacy-Preserving Techniques for Federated Analysis
| Feature | Differential Privacy (DP) | Homomorphic Encryption (HE) | Hybrid Approach (PPML-Hybrid) |
|---|---|---|---|
| Privacy Basis | Mathematical guarantee via calibrated noise [64] [67]. | Cryptographic security via encryption [67]. | Combines both DP and HE. |
| Impact on Utility | Can reduce model accuracy due to noise [67]. | Preserves model accuracy (noise-free) [67]. | Balances utility; more HE clients can improve accuracy [67]. |
| Computational Cost | Low [67]. | High [67]. | Flexible; adapts to client resources [67]. |
| Best For | Scenarios with limited compute or where formal, mathematical privacy guarantees are required. | Scenarios where model accuracy is critical and sufficient computational resources are available. | Heterogeneous environments with varying client capabilities and privacy needs [67]. |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Components for a Federated Analysis Platform
| Item | Function |
|---|---|
| Federated Database Management System (FDBMS) | The central software that receives global queries, breaks them into sub-queries, orchestrates execution across nodes, and reassembles the results [63]. |
| Common Data Model (e.g., OMOP) | A standardized data schema that ensures semantic interoperability, meaning that the same data element (e.g., a diagnosis) is represented consistently across all data partners [65]. |
| Data Connectors | Lightweight software agents installed at each data source that enable the FDBMS to communicate securely with diverse local data systems (e.g., SQL databases, data lakes) [63]. |
| Differential Privacy Library (e.g., TensorFlow Privacy) | A software library that provides algorithms for adding calibrated noise to data or model updates to achieve a mathematically rigorous privacy guarantee [64]. |
| Homomorphic Encryption Library (e.g., Microsoft SEAL) | A software library that implements encryption schemes (like CKKS) allowing computation on encrypted data, enabling secure aggregation in federated learning [67]. |
| Data Catalog & Metadata Repository | A searchable central inventory containing metadata (data about the data), making distributed datasets findable and understandable for researchers without exposing raw data [63]. |
Ensuring Robustness: Benchmarking, Clinical Application, and Impact Assessment
Establishing Gold Standards for Benchmarking Integration Methodologies
Frequently Asked Questions
FAQ 1: What are the main categories of single-cell multimodal omics data integration, and why is this categorization important for benchmarking?
The systematic categorization of integration methods is foundational for meaningful benchmarking. Based on input data structure and modality combination, methods are defined into four prototypical categories [70]:
- Vertical Integration: For analyzing multiple modalities (e.g., RNA, ADT, ATAC) profiled from the same set of cells.
- Diagonal Integration: For integrating datasets that profile different, but overlapping, sets of modalities.
- Mosaic Integration: For integrating data from different batches or conditions where each batch may have a different combination of modalities.
- Cross Integration: For integrating data across different biological samples or studies.
This categorization is crucial because a method's performance is highly dependent on the data structure and modality combination it is applied to. Benchmarking studies evaluate methods separately for each category to provide fair and actionable guidance [70].
FAQ 2: My integrated data shows poor separation of known cell types after applying a vertical integration method. What could be the issue?
Poor biological preservation after integration can stem from several issues. The benchmarking study identified that method performance is both dataset-dependent and, more notably, modality-dependent [70]. To troubleshoot:
- Verify Method Suitability: Ensure the method you've chosen is designed for your specific modality combination (e.g., RNA+ADT, RNA+ATAC). Consult benchmarking results to select a method that has been shown to perform well for dimension reduction and clustering on your data type. For instance, on a representative RNA+ADT dataset, methods like Seurat WNN, sciPENN, and Multigrate demonstrated generally better performance in preserving biological variation [70].
- Check Evaluation Metrics: Use multiple metrics to diagnose the problem. A method might rank highly by one metric (e.g., iF1, NMIcellType) but poorly by another (e.g., ASWcellType, iASW), indicating a trade-off between clustering accuracy and continuous manifold preservation [70].
- Inspect Input Data: Confirm that your raw data quality is high and that pre-processing (e.g., normalization, filtering) was appropriate for all modalities.
FAQ 3: How can I reliably identify molecular markers from my multimodal data for cell type annotation?
Only a subset of vertical integration methods, such as Matilda, scMoMaT, and MOFA+, support feature selection [70]. The troubleshooting steps below outline their key differences and how to evaluate their output.
- Diagnosis: Determine whether you need cell-type-specific markers or a general set of informative features.
- Solution:
- Use Matilda or scMoMaT to identify distinct molecular markers for each cell type in your dataset.
- Use MOFA+ to select a single, cell-type-invariant set of markers that are informative across the entire dataset.
- Validation: Benchmarking shows that features selected by scMoMaT and Matilda generally lead to better clustering and classification of cell types. In contrast, MOFA+ may generate more reproducible feature selection results across different data modalities [70]. Validate the selected markers against known biology.
Troubleshooting Guides
Issue 1: Selecting an Inappropriate Integration Method for the Task and Data
Problem: A method is chosen without consideration for the specific integration category (vertical, diagonal, mosaic, cross) or the computational task (dimension reduction, batch correction, feature selection, etc.), leading to suboptimal or incorrect results [70].
Investigation Protocol:
- Categorize Your Data: Classify your dataset according to the four integration categories (see FAQ 1).
- Define Your Primary Task: Identify the key goal of your analysis (e.g., cell type clustering, batch correction, marker identification).
- Consult Benchmarking Tables: Refer to published benchmarking studies, like the one cited, which provide performance rankings of methods by category and task [70].
Resolution Steps:
- Method Selection: Based on your data category and task, select a top-performing method from the relevant benchmark.
- Example: For vertical integration for dimension reduction and clustering on RNA+ADT data, high-performing methods include Seurat WNN, Multigrate, and sciPENN [70].
- Parameter Tuning: Use the method's documentation to optimize key parameters for your specific dataset.
Issue 2: Poor Performance in Batch Correction or Data Harmonization
Problem: Technical batch effects are not adequately removed during integration, confounding biological signals. This is a common challenge in multi-omics data harmonization [31].
Investigation Protocol:
- Visual Inspection: Use UMAP or t-SNE plots colored by batch to see if batches remain separate after integration.
- Metric Calculation: Quantify batch correction using metrics like iLISI (integration Local Inverse Simpson's Index) or other batch mixing metrics employed in benchmarks [70].
- Biological Preservation Check: Ensure that desired biological variation (e.g., cell types) has not been over-corrected. Calculate metrics like ASW_cellType (Average Silhouette Width for cell type).
Resolution Steps:
- Apply Dedicated Methods: For complex diagonal or mosaic integration scenarios, ensure you are using a method designed for these tasks, as their ability to handle varying modality combinations inherently involves batch correction [70].
- Leverage AI Techniques: For large-scale multi-omics data, consider AI-driven methods. Tools like ComBat can be used for batch correction, and deep learning models are increasingly effective for non-linear batch effect removal and data harmonization [31].
- Iterative Refinement: If biological signals are lost, try a different method or adjust the strength of the batch correction parameter, if available.
Experimental Protocols & Data
Protocol 1: Benchmarking an Integration Method for Dimension Reduction and Clustering
This protocol outlines the procedure used in large-scale benchmarking studies to evaluate method performance [70].
1. Objective: Systematically evaluate and compare the performance of single-cell multimodal omics integration methods on dimension reduction and clustering tasks.
2. Materials and Reagents
| Item | Function in Experiment |
|---|---|
| Real Single-Cell Multimodal Datasets (e.g., CITE-seq, SHARE-seq) | Provide a ground-truth biological context with known cell types for evaluating biological preservation. |
| Simulated Datasets | Allow for evaluation under controlled conditions where the true data structure is known. |
| Computational Infrastructure (High-performance computing cluster) | Enables the running of multiple computationally intensive integration methods. |
| Evaluation Metric Suite (e.g., ASW_cellType, iF1, NMI) | Quantifies different aspects of method performance (clustering accuracy, batch mixing, etc.). |
3. Methodology
- Data Curation: Assemble a panel of real and simulated datasets covering various modality combinations (RNA+ADT, RNA+ATAC, RNA+ADT+ATAC).
- Method Application: Run each applicable integration method on each dataset according to its standard workflow.
- Output Extraction: Obtain a low-dimensional embedding or a graph from each method.
- Performance Quantification: Calculate a panel of pre-defined evaluation metrics on the outputs. For dimension reduction, this may include ASW_cellType. For clustering, metrics like iF1 and NMI are used [70].
- Rank Aggregation: Summarize performance across all datasets and metrics to compute an overall rank score for each method.
4. Expected Output: A ranked list of integration methods for each data modality combination and task, providing a data-driven guideline for method selection.
Quantitative Benchmarking Data
The table below summarizes the grand rank scores of top-performing vertical integration methods from a comprehensive benchmark, illustrating how performance varies by data modality [70].
Table 1: Performance of Vertical Integration Methods by Data Modality
| Method | RNA + ADT Grand Rank | RNA + ATAC Grand Rank | RNA + ADT + ATAC Grand Rank |
|---|---|---|---|
| Seurat WNN | 1 | 2 | - |
| Multigrate | 2 | 4 | 1 |
| sciPENN | 3 | - | - |
| UnitedNet | - | 1 | - |
| Matilda | 4 | 3 | 2 |
| ... other methods ... | ... | ... | ... |
Note: A lower rank score indicates better overall performance. Dashes indicate the method was not among the top performers for that modality or was not applicable. Performance is dataset-dependent; this table provides a summary guide.
The Scientist's Toolkit
Table 2: Key Reagents and Computational Tools for Multimodal Integration
| Item | Category | Function |
|---|---|---|
| CITE-seq Data | Biological Data | A common source of paired RNA and protein abundance (ADT) data for benchmarking vertical integration [70]. |
| SHARE-seq Data | Biological Data | Provides paired RNA and ATAC-seq data from the same single cell for benchmarking [70]. |
| Seurat WNN | Software/Method | A top-performing method for vertical integration, particularly on RNA+ADT data. It uses a weighted nearest neighbor approach to combine modalities [70]. |
| Multigrate | Software/Method | A top-performing method for vertical integration across multiple modalities (RNA+ADT, RNA+ATAC, trimodal). It creates a joint generative model of the data [70]. |
| MOFA+ | Software/Method | A factor analysis model that is effective for multi-group integration and can perform feature selection [70]. |
| ComBat | Software/Tool | A widely used algorithm for adjusting for batch effects in high-dimensional genomic data, often employed in data harmonization [31]. |
| Graph Neural Networks (GNNs) | AI Methodology | A cutting-edge AI approach used to model biological networks (e.g., protein-protein interactions) perturbed by mutations, aiding in multi-omics interpretation [31]. |
Workflow and Relationship Visualizations
Decision Framework for Integration Method Selection
Multi-omics Integration and Benchmarking Workflow
Multi-omics approaches integrate diverse biological data layers—including genomics, transcriptomics, proteomics, and metabolomics—to create a comprehensive understanding of health and disease. Data harmonization is the critical process of standardizing and integrating these disparate datasets to ensure compatibility, comparability, and reproducibility. This technical support center provides troubleshooting guidance and best practices for overcoming key challenges in multi-omics research, framed within the context of a broader thesis on data harmonization best practices.
Frequently Asked Questions (FAQs)
Q1: Why is data harmonization considered the foundation of reliable multi-omics analysis?
Data harmonization addresses the fundamental challenge of data heterogeneity. Each omics discipline generates massive datasets with unique formats, measurement technologies, and analytical methods. Without harmonization, technical variations and biases obscure true biological signals, compromising the accuracy and reproducibility of integrated analyses [6]. Harmonization through standardized protocols and quality control ensures that results are reliable and comparable across different studies and platforms [6].
Q2: What are the primary strategies for integrating multiple omics datasets?
Researchers typically employ three main integration strategies, each with distinct advantages and challenges [12]:
- Early Integration: Merges all raw features from different omics layers into a single dataset before analysis. Best for capturing all potential cross-omics interactions but computationally intensive and prone to the "curse of dimensionality."
- Intermediate Integration: First transforms each omics dataset into a lower-dimensional or network-based representation before combination. Effectively reduces complexity and can incorporate biological context.
- Late Integration: Analyzes each omics type separately and combines the results or predictions at the final stage. Handles missing data well and is computationally efficient, but may miss subtle interactions between different data layers.
Q3: How can batch effects be identified and corrected in multi-omics studies?
Batch effects—systematic technical biases introduced by different reagents, technicians, or sequencing machines—are a major concern. They can be identified through Principal Component Analysis (PCA) and other visualization tools, where samples may cluster by batch rather than biological group. Correction methods include specialized statistical tools like ComBat, which standardizes data across batches, and careful experimental design that randomizes samples across processing batches [12].
Q4: What is the role of AI and machine learning in multi-omics data harmonization and analysis?
AI and machine learning are indispensable for handling the scale and complexity of multi-omics data [5] [12] [71]. They act as advanced tools for pattern recognition, capable of detecting subtle connections across millions of data points. Key applications include:
- Dimensionality Reduction: Autoencoders compress high-dimensional data into manageable latent spaces.
- Network Integration: Graph Convolutional Networks learn from biological networks to make predictions.
- Data Fusion: Similarity Network Fusion creates and fuses patient-similarity networks from different omics layers.
Q5: What are the best practices for validating a multi-omics biomarker signature for clinical use?
Robust validation is essential for clinical translation. Key practices include [72]:
- Rigorous Statistical Validation: Employ strict cross-validation and hold-out validation methods to prevent overfitting.
- Multicohort Validation: Test the biomarker signature on independent, external cohorts to ensure generalizability.
- Clinical Utility Assessment: Demonstrate that the biomarker provides actionable information that improves patient outcomes, diagnosis, or treatment selection.
Troubleshooting Common Multi-Omics Workflow Issues
Table 1: Common Data Harmonization Challenges and Solutions
| Challenge | Symptom | Root Cause | Solution |
|---|---|---|---|
| Data Heterogeneity | Inability to merge datasets; inconsistent results. | Different data formats, scales, and technological platforms [12]. | Implement standardized data formats (e.g., standardized file formats like .mzML for proteomics) and ontologies; use data harmonization software [6]. |
| Missing Data | Incomplete datasets bias analysis and reduce statistical power. | Sample limitations, analytical dropouts, or cost constraints [12]. | Apply robust imputation methods (e.g., k-nearest neighbors) or use analysis models (like late integration) that can handle missing data types [12]. |
| Batch Effects | Samples cluster by processing date or batch instead of biological group. | Technical variations from different processing runs, reagents, or personnel [12]. | Use batch correction algorithms (e.g., ComBat); randomize samples across batches during experimental design [12]. |
| Low Statistical Power | Failure to replicate findings; inability to detect significant signals. | Insufficient sample size relative to the high number of features analyzed ("curse of dimensionality") [72]. | Ensure adequate sample size through power analysis; collaborate to pool cohorts; apply stringent statistical filters [72]. |
| Poor Clinical Translation | A biomarker model performs well in discovery but fails in independent validation. | Overfitting during discovery phase; lack of biological relevance; cohort-specific biases [72]. | Apply strict filtering; integrate prior biological knowledge; validate across multiple, diverse cohorts [72] [73]. |
Experimental Protocols for Data Harmonization
Protocol 1: Preprocessing and Normalization of Multi-Omics Data
Objective: To transform raw data from various omics platforms into a normalized and comparable format ready for integrated analysis.
Materials:
- Computing Environment: High-performance computing or cloud-based infrastructure (e.g., AWS, Google Cloud).
- Software Tools: R/Python with packages for omics analysis (e.g.,
limma,DESeq2for RNA-seq;SWATH2statsfor proteomics).
Methodology:
- Quality Control: For each dataset, perform platform-specific QC (e.g., check sequencing depth and alignment rates for genomics; assess mass spectrometry peak intensity and retention time for proteomics).
- Normalization: Apply data-type-specific normalization to remove technical variations.
- Transcriptomics (RNA-seq): Use methods like TPM (Transcripts Per Million) or FPKM (Fragments Per Kilobase Million) to correct for sequencing depth and gene length [12].
- Proteomics: Apply intensity normalization or variance-stabilizing transformation to mass spectrometry data [12].
- Genomics: For gene variant data, focus on quality and depth filters.
- Batch Effect Correction: Use statistical methods like ComBat or surrogate variable analysis (SVA) to adjust for non-biological technical variance [12].
- Data Annotation: Map all features (e.g., genes, proteins) to standardized biological identifiers (e.g., Ensembl IDs, UniProt IDs) and pathways (e.g., KEGG, Reactome) to enable cross-omics integration.
Protocol 2: Network-Based Integration for Biomarker Discovery
Objective: To identify robust, biologically grounded biomarker signatures by integrating multi-omics data onto shared biochemical networks.
Materials:
- Prior Knowledge Databases: Protein-protein interaction databases (e.g., STRING), pathway databases (e.g., KEGG, Reactome).
- Analysis Tools: Network analysis software (e.g., Cytoscape) or dedicated multi-omics integrators (e.g., OmicsIntegrator) [6].
Methodology:
- Network Construction: For each omics layer, construct a network. For example, create a gene co-expression network from transcriptomics data or a protein-protein interaction network from proteomics data.
- Data Mapping: Map differentially expressed genes, proteins, or metabolites from your analysis onto the integrated network.
- Module Identification: Use network algorithms to identify densely connected "modules" or sub-networks that are enriched with features from multiple omics layers. These modules often represent key functional units or pathways dysregulated in the disease state [5].
- Biomarker Prioritization: Prioritize features (biomarker candidates) that are central (hubs) within these cross-omics modules, as they are more likely to be functionally important and yield robust signatures [5] [73].
The following diagram illustrates this network-based integration workflow.
Protocol 3: AI-Driven Patient Stratification Using Multi-Omics Data
Objective: To use machine learning to identify distinct patient subgroups based on integrated multi-omics profiles.
Materials:
- Data: A harmonized multi-omics dataset with linked clinical outcomes.
- Software: Python/R with ML libraries (e.g.,
scikit-learn,PyTorch).
Methodology:
- Feature Selection: Reduce dimensionality by selecting the most informative features from the integrated dataset using methods like variance filtering or univariate statistical tests.
- Model Building: Apply unsupervised learning algorithms to discover inherent patient clusters.
- Clustering: Use methods like k-means or hierarchical clustering on the multi-omics data.
- Similarity Network Fusion (SNF): Fuse patient-similarity networks from each omics layer into a single network and then perform clustering on this fused network to identify robust patient subgroups [12].
- Subgroup Characterization: Statistically compare the clinical outcomes (e.g., survival, drug response) and molecular profiles of the identified subgroups to validate their clinical relevance.
- Biomarker Extraction: Identify the key molecular features (e.g., specific genes, proteins) that most strongly define each subgroup. These form the basis for a stratification biomarker test [73] [71].
Essential Research Reagent Solutions
Table 2: Key Research Reagents and Materials for Multi-Omics Studies
| Item | Function in Multi-Omics Research | Application Example |
|---|---|---|
| Next-Generation Sequencing (NGS) Kits | For generating genomic (DNA) and transcriptomic (RNA) data from patient samples. | Whole genome sequencing to identify genetic variants; RNA-seq for gene expression profiling [5] [12]. |
| Mass Spectrometry Kits & Reagents | For quantifying proteins (proteomics) and small molecules (metabolomics). | Profiling the proteome of tumor tissues to identify differentially expressed proteins and potential drug targets [12]. |
| Single-Cell Isolation Kits | To separate individual cells for high-resolution omics profiling. | Single-cell RNA sequencing to understand cellular heterogeneity within a tumor and identify rare cell populations [5]. |
| Liquid Biopsy Collection Tubes | For stable isolation of cell-free DNA (cfDNA), RNA, and proteins from blood samples. | Isolating circulating tumor DNA (ctDNA) for non-invasive cancer detection and monitoring treatment response [5] [6]. |
| Multi-Omics Data Integration Software | Computational platforms and pipelines for harmonizing and analyzing diverse omics datasets. | Tools like OmicsIntegrator are used for network-based integration of genomic, transcriptomic, and proteomic data [6]. |
Multi-Omics Data Harmonization and Analysis Workflow
The following diagram provides a high-level overview of the end-to-end process for harmonizing and analyzing multi-omics data, from raw data to clinical insight.
Core Concepts: The "What" and "Why" of Multi-Omics Harmonization
What is multi-omics data harmonization? Multi-omics data harmonization is the process of bringing data from different molecular layers—such as genomics, transcriptomics, proteomics, and metabolomics—into a compatible and standardized format. This enables their joint analysis to form a unified biological picture. It involves steps like data curation, ID mapping, quality control, and normalization to account for differences in measurement units, scales, and technical biases across platforms [74] [15].
Why is harmonization critical in oncology and neurodegenerative disease research? Complex diseases like cancer and neurodegenerative disorders involve intricate interactions across multiple molecular layers. Harmonization is crucial because it enables researchers to move beyond a siloed view and capture the full complexity of these diseases.
- In Oncology, an integrated view can identify novel biomarkers and therapeutic targets. For example, integrating genomic and proteomic data (proteogenomics) has refined the prediction of therapeutic responses [75].
- In Neurodegenerative Diseases, multi-omics integration helps decipher complex genetic architectures, addressing challenges like "missing heritability" and revealing the molecular mechanisms behind diseases like Alzheimer's and Parkinson's [76].
Troubleshooting Guide: Common Multi-Omics Integration Pitfalls and Solutions
This guide addresses frequent technical challenges encountered during multi-omics data integration.
| Pitfall | Underlying Problem | Recommended Solution |
|---|---|---|
| Unmatched Samples | Data from different sample sets or patients are forced together, confusing results [77]. | Create a sample matching matrix; analyze only paired samples or use meta-analysis models [77]. |
| Misaligned Resolution | Incompatible data resolutions (e.g., bulk vs. single-cell) lead to misleading correlations [77]. | Use reference-based deconvolution for bulk data or define shared integration anchors for single-cell data [77]. |
| Improper Normalization | Different normalization methods per modality (e.g., TPM for RNA, β-values for methylation) bias integration [15] [77]. | Apply comparable scaling (e.g., log transformation, Z-scoring, quantile normalization) to all layers [77]. |
| Ignoring Batch Effects | Batch effects from different processing labs compound across layers, creating false biological signals [77]. | Inspect batch structure across layers; apply cross-modal batch correction (e.g., Harmony) with biological covariates [77]. |
| Overinterpreting Weak Correlations | Assuming mRNA-protein correlation is high; building networks from biologically weak associations [77]. | Only analyze regulatory links supported by mechanistic logic (e.g., distance, motif analysis); report confidence levels [77]. |
Frequently Asked Questions (FAQs)
Q1: We have RNA-seq and proteomics data from overlapping but not identical patient sets. Can we still integrate them? Yes, but with caution. Forcing unpaired data will likely produce noise. Instead, stratify your analysis:
- Perform a primary analysis only on the perfectly matched samples.
- For the remaining unpaired data, use group-level summarization or meta-analysis models with clear documentation of the limitations [77].
Q2: Our integrated analysis is dominated by signals from one data type (e.g., ATAC-seq), drowning out others. What went wrong? This is typically a normalization or scaling issue. Different data types have different native scales and variances. If one modality (like raw ATAC-seq counts) is not normalized while others are, it will dominate variance-based analyses like PCA.
- Solution: Ensure each omics layer is brought to a comparable scale using appropriate transformations (e.g., log, CLR, quantile normalization) before integration [77].
Q3: Why is there often a poor correlation between mRNA expression and protein abundance in our integrated datasets? A weak mRNA-protein correlation is a common biological reality, not necessarily an analysis error. Protein levels are influenced by post-transcriptional regulation, translation rates, and protein degradation.
- Solution: Do not assume high correlation. Treat discordant signals as biologically informative, suggesting potential post-transcriptional regulation. Focus on pathway-level coherence rather than individual gene-protein pairs [77].
Q4: What is the single most important step for a successful multi-omics integration project? The most critical step is project design from the user's perspective. Before starting, define real use-case scenarios and pretend you are the end-user analyst. This ensures the final integrated resource is functional, interpretable, and addresses genuine biological questions, rather than being optimized only for the data curators [15].
Experimental Protocols for Data Harmonization
Protocol 1: Standardized Data Preprocessing and Metadata Collection
Objective: To transform raw data from diverse omics platforms into a harmonized, analysis-ready format.
Materials:
- Raw Data: FASTQ, .idat, or vendor-specific mass spec files.
- Computing Environment: Unix command line, R or Python.
- Reference Databases: ENSEMBL, UniProt, HMDB for ID mapping.
Methodology:
- Data Standardization: Convert all data into a unified format, such as an n-by-k samples-by-feature matrix. This often involves:
- Genomics/Transcriptomics: Alignment to a reference genome, gene count quantification, and normalization (e.g., TPM, FPKM).
- Proteomics: Peak identification, protein quantification, and normalization by spectral counts or TMT ratios.
- Methylomics: Calculation of β-values or M-values for each CpG site [15] [77].
- ID Mapping: Map all features (e.g., genes, proteins, metabolites) to a common set of standard identifiers (e.g., ENSEMBL IDs, HGNC symbols) to ensure features can be linked across datasets [74].
- Metadata Annotation: For every sample, collect rich metadata, including sample origin, processing protocol, batch information, and donor clinical data. This is as crucial as the molecular data itself [15].
Protocol 2: Multi-Omics Factor Analysis (MOFA) for Data Integration
Objective: To identify the principal sources of variation (factors) across multiple omics datasets.
Materials:
- Input Data: Harmonized and preprocessed data matrices from at least two omics layers.
- Software/Tool: MOFA+ (R/Python package) [74].
Methodology:
- Input Preparation: Format your preprocessed and normalized omics data into the sample-by-feature matrices required by MOFA+. Ensure sample names are aligned across matrices.
- Model Training: Run MOFA+ to decompose the variation in the data. The model will infer a set of factors that are shared across data types as well as factors specific to individual data types.
- Result Interpretation:
- Factor Analysis: Examine the factor scores to understand which factors capture key biological or technical patterns (e.g., disease status, batch effects).
- Weight Inspection: For each factor, analyze the feature weights to identify which genes, proteins, or metabolites are driving the pattern [74].
Visualization of Workflows and Relationships
Multi-Omics Integration Workflow
Data Harmonization Logic
| Tool / Resource | Function | Application Context |
|---|---|---|
| Flexynesis | A deep learning toolkit that streamlines data processing, feature selection, and model training for bulk multi-omics data. | Accessible multi-omics integration for precision oncology tasks like drug response prediction and survival modeling [78]. |
| Cytoscape | An open-source platform for visualizing complex molecular interaction networks and integrating these with other data types. | Visualizing integrated networks to identify key subnetworks or hubs associated with a disease phenotype [74]. |
| MOFA+ | A statistical tool for multi-omics factor analysis that discovers the principal sources of variation across multiple data modalities. | Uncovering shared and specific patterns of variation across omics layers in an unsupervised manner [74]. |
| TCGA/CCLE | Publicly available databases containing comprehensive molecular profiling data for thousands of tumor samples and cancer cell lines. | Benchmarking integration methods, discovering biomarkers, and understanding cancer biology [75] [78]. |
| Unix Command Line & R | Computational environments essential for running preprocessing, normalization, and integration scripts. | Required for most data harmonization and analysis workflows; basic proficiency is necessary [74]. |
Comparative Analysis of Integration Tools and Their Performance Metrics
In multi-omics studies, the integration of data from genomics, transcriptomics, proteomics, and metabolomics is essential for uncovering complex biological relationships [44]. However, this integration presents significant computational challenges due to data heterogeneity, varying measurement units, and technical noise [15] [79]. This technical support center provides troubleshooting guides and FAQs to help researchers navigate these challenges, framed within best practices for data harmonization in multi-omics research.
Performance Metrics and Tool Comparison
Key Performance Metrics for Multi-Omics Integration
The table below summarizes essential metrics for evaluating multi-omics integration tools, derived from benchmark studies [79] [80].
| Metric Category | Specific Metric | Optimal Range/Value | Interpretation in Multi-Omics Context |
|---|---|---|---|
| Clustering Performance | Adjusted Rand Index (ARI) | Higher value (0-1) | Measures sample clustering accuracy against known biological groups [79]. |
| Survival Difference (Log-rank test) | p-value < 0.05 | Indicates whether identified clusters have significant clinical relevance [79]. | |
| Data Quality & Reproducibility | Signal-to-Noise Ratio (SNR) | Higher value | Assesses the ratio of true biological signal to technical noise; crucial for ratio-based profiling [80]. |
| Mendelian Concordance Rate | > 99% | For family-based designs, measures genotyping accuracy [80]. | |
| Technical Robustness | Batch Effect Correction | No vendor/lab clustering in PCA | Evaluates the tool's ability to remove non-biological technical variations [77] [80]. |
| Performance under Noise | ARI reduction < 30% with 30% added noise | Tests the robustness of the integration method when noise levels are high [79]. |
Comparative Analysis of Multi-Omics Integration Tools
The following table compares the performance and characteristics of various tools and approaches used for multi-omics data integration, based on recent benchmarking studies and literature [78] [79] [44].
| Tool/Method | Primary Approach | Best Suited Omics Types | Reported Performance/Strengths | Key Limitations |
|---|---|---|---|---|
| Flexynesis [78] | Deep Learning (DL) | Bulk transcriptomics, genomics, epigenomics | High accuracy (AUC=0.981) for MSI status classification; supports multi-task learning. | Requires medium-to-large sample sizes; complex hyperparameter tuning. |
| MOFA+ [44] | Factor Analysis | Multiple (Transcriptomics, Proteomics, Metabolomics) | Identifies latent factors driving variation across omics layers; good for exploratory analysis. | Can miss modality-specific signals; requires careful interpretation. |
| WGCNA [44] | Correlation Network Analysis | Transcriptomics, Proteomics, Metabolomics | Identifies modules of highly correlated features (genes/proteins/metabolites). | Primarily for pairwise integration; limited to linear relationships. |
| xMWAS [44] | Multivariate Association | Multiple (Transcriptomics, Proteomics, Metabolomics) | Builds integrative networks and identifies communities of interconnected features. | Association does not imply causation; requires significance thresholds. |
| Simple Correlation [44] | Statistical Correlation | Proteomics, Metabolomics, Transcriptomics | Easy to implement and interpret (e.g., scatter plots, Pearson/Spearman correlation). | Can only capture linear, pairwise relationships; prone to false positives. |
| DIABLO [77] | Multivariate (sPLS-DA) | Multiple (Transcriptomics, Proteomics, Metabolomics) | Effective for supervised classification and biomarker discovery; handles multiple datasets. | Performance can degrade with high dimensionality and low sample size. |
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function in Multi-Omics Integration |
|---|---|
| Quartet Reference Materials [80] | Provides multi-omics ground truth from matched DNA, RNA, protein, and metabolites derived from a family quartet for objective QC and method benchmarking. |
| Common Data Model (CDM) [81] | A universal schema or "lingua franca" that standardizes data structure, naming conventions, and definitions, enabling semantic alignment across disparate datasets. |
| Controlled Vocabularies & Ontologies (e.g., SNOMED CT, GO) [81] | Formal representations of knowledge with defined concepts and relationships, ensuring that data from different sources is harmonized with consistent meaning. |
| Batch Effect Correction Algorithms (e.g., ComBat) [81] | Statistical methods to identify and remove technical noise introduced when samples are processed in different batches or on different days. |
Troubleshooting Guides and FAQs
FAQ 1: Our integrated multi-omics data shows poor correlation between mRNA and protein levels for our candidate biomarkers. Is this a technical error?
Answer: Not necessarily. A weak correlation between mRNA and protein is a common biological phenomenon, not always a technical flaw [77].
- Potential Cause 1: Biological Regulation. Post-transcriptional regulation (e.g., miRNA), protein degradation rates, and translational efficiency can decouple mRNA abundance from protein levels.
- Solution: Do not assume high correlation. Use prior knowledge of regulatory mechanisms (e.g., miRNA targets) to inform your interpretation. Focus on protein-level data for downstream biomarker validation if that is the functional layer of interest.
- Potential Cause 2: Unmatched Samples or Time Points. The RNA and protein data may come from different subsets of patients or be measured at different time points in a dynamic process [77].
- Solution: Create a sample matching matrix to visualize the true overlap between omics layers. For temporal studies, align all measurements to a common timeline before integration.
FAQ 2: When we integrate our ATAC-seq and RNA-seq data, the clustering results are completely dominated by the ATAC-seq signal. What went wrong?
Answer: This is typically caused by improper normalization across the different data modalities [77].
- Root Cause: Each omics type has unique statistical characteristics. If one dataset (e.g., ATAC-seq raw counts) is not normalized while others are (e.g., RNA-seq Z-scores), the unnormalized data with higher inherent variance will dominate multivariate analyses like PCA.
- Solution:
- Apply modality-specific scaling: Normalize each omics layer individually to make their distributions comparable. This may include log-transformation for sequencing data, centered log-ratio (CLR) for compositional data, or Z-scoring.
- Use integration-aware tools: Employ methods like MOFA+ or DIABLO that are explicitly designed to weight different modalities appropriately, rather than simply concatenating datasets and running PCA [77].
FAQ 3: Our multi-omics clusters separate samples by sequencing batch, not by biological condition. How can we correct for this?
Answer: This indicates a strong batch effect that must be addressed before biological interpretation [77] [80].
- Solution Strategy:
- Horizontal Integration First: Apply batch effect correction methods (e.g., ComBat, Harmony) to each omics type individually (e.g., correct all RNA-seq data together) before attempting cross-omics (vertical) integration [80].
- Cross-modal Batch Alignment: After within-omics correction, check if a sample's position in one omics space is consistent with its position in another. If not, apply cross-modal batch correction.
- Leverage Reference Materials: Use multi-omics reference materials (like the Quartet samples) measured across all your batches to objectively quantify and correct for batch effects [80].
- Critical Check: Always visualize your data using PCA after correction to confirm that the primary principal components are driven by biology, not technical artifacts.
FAQ 4: We have a small sample size (n < 20). Which integration tools are most suitable?
Answer: Small sample sizes and high dimensionality are a major challenge. Your tool choice is critical.
- Recommendations:
- Aggressive Feature Selection: Reduce dimensionality drastically before integration. Select less than 10% of omics features, focusing on those with known biological relevance to your study to improve clustering performance by up to 34% [79].
- Avoid Complex Deep Learning Models: Tools like Flexynesis, which are powerful for large datasets, may overfit on small sample sizes [78].
- Prefer Classical/Multivariate Methods: Consider methods like DIABLO or MOFA+, which can be more robust in low-sample settings, provided feature selection is applied [77] [44].
- Utilize Ratio-based Profiling: If possible, use a ratio-based approach (e.g., scaling all samples to a common reference) to improve data comparability and reproducibility with small n [80].
Experimental Protocols and Workflows
Standard Protocol for Multi-Omics Data Harmonization
The following workflow outlines a robust, step-by-step procedure for harmonizing multi-omics data, incorporating best practices for preprocessing and integration [15] [81] [80].
Multi-Omics Harmonization Workflow
Protocol Steps:
- Data Discovery & Profiling: Create a comprehensive inventory of all data sources. Perform deep-dive analysis to understand data structures, value ranges, and quality issues (e.g., nulls, duplicates) [81].
- Define a Common Data Model (CDM): Establish a target schema or "lingua franca" for your data. This includes standardized naming conventions and a data dictionary to ensure semantic alignment (e.g., defining "patient_age" uniformly) [81].
- Preprocessing & Standardization (Per Omics Type):
- Normalization: Apply modality-specific normalization (e.g., TPM for RNA-seq, β-values for methylation, spectral count normalization for proteomics) to account for differences in measurement units and distributions [15] [77].
- Handle Missing Values: Use appropriate imputation methods or removal based on the extent and nature of the missing data.
- Feature Selection: Aggressively filter features to reduce dimensionality. Use biological knowledge (e.g., known pathways) or statistical measures (e.g., variance) to select less than 10% of total features, which has been shown to significantly improve clustering performance [79].
- Horizontal Integration (Within-Omics Batch Correction): Integrate datasets from the same omics type generated across different batches, labs, or platforms. Use methods like ComBat or Harmony to remove technical batch effects while preserving biological variation. The Quartet Project's ratio-based profiling (scaling samples to a common reference) is a highly effective strategy for this step [80].
- Vertical Integration (Cross-Omics Integration): Combine the harmonized datasets from different omics layers. Choose a method based on your biological question:
- Validation & Biological Interpretation: Validate the integration results against ground truth if available (e.g., Quartet family relationships) [80]. Use functional enrichment analysis on identified modules or features. Crucially, explicitly analyze and interpret both shared and modality-specific signals, as the discordance can be biologically informative [77].
Protocol for Implementing the Quartet Project's Ratio-Based Profiling
The Quartet Project provides a robust framework for assessing and improving multi-omics integration using reference materials from a family quartet. The core innovation is ratio-based profiling to enhance reproducibility [80].
Quartet Ratio-Based Profiling Protocol
Experimental Steps:
- Concurrent Measurement: For each omics assay (e.g., RNA-seq, proteomics), process your study samples alongside aliquots of the Quartet reference materials (samples D5, D6, F7, M8) in the same batch [80].
- Ratio-Based Calculation: On a feature-by-feature basis (e.g., per gene, per protein), calculate a ratio by scaling the absolute feature value of a study sample relative to the value of a designated common reference sample (e.g., D6). This transforms "absolute" quantification into a relative measurement that is more reproducible across labs and platforms [80].
- Horizontal Integration QC: Use the built-in truths of the Quartet materials to quality-check each omics data type.
- Calculate the Signal-to-Noise Ratio (SNR) to evaluate the precision of quantitative measurements.
- For genomic data, calculate the Mendelian Concordance Rate to evaluate genotyping accuracy [80].
- Vertical Integration & Final QC: After integrating the ratio-based data from multiple omics layers, perform two critical QC checks:
- Sample Clustering QC: Verify that the integrated data can correctly classify the four Quartet individuals and also group the monozygotic twins (D5, D6) together, resulting in three genetically distinct clusters [80].
- Central Dogma QC: Assess whether the integrated analysis can recapitulate the fundamental biological flow of information from DNA to RNA to Protein for known relationships [80].
Frequently Asked Questions (FAQs)
1. What is the core difference between data integration and data harmonization? While often used interchangeably, these terms describe different processes. Data integration combines data from various sources into a single, accessible location. In contrast, data harmonization is the process of standardizing and converting fragmented data from multiple sources into a unified, comparable format by resolving differences in syntax (formats), structure (schemas), and semantics (meaning). Harmonization ensures that data means the same thing everywhere, which is a critical prerequisite for meaningful integration and analysis [82].
2. Why are my multi-omics datasets so difficult to correlate and analyze? Multi-omics data integration is challenging due to several inherent factors [2] [8]:
- Heterogeneity: Each omics type (genomics, transcriptomics, proteomics, etc.) has unique data structures, statistical distributions, and noise profiles.
- High Dimensionality: The number of variables (e.g., genes, proteins) vastly exceeds the number of samples, a problem known as High Dimension Low Sample Size (HDLSS), which can cause machine learning models to overfit [8].
- Technical Variation: Batch effects from different technicians, reagents, or sequencing machines can create systematic noise that obscures real biological signals [12].
- Missing Data: It is common for a sample to have data for one omics layer but not another, which can bias analysis if not handled with robust imputation methods [12].
3. What are the primary strategies for integrating multiple omics datasets? Integration strategies are often categorized by when the combination of datasets occurs [12] [8]:
- Early Integration: All omics datasets are merged into a single large matrix before analysis. This can capture all interactions but results in a highly complex and noisy dataset [12] [8].
- Intermediate Integration: Datasets are first transformed into new representations (e.g., biological networks) and then combined. This reduces complexity and can incorporate biological context [12].
- Late Integration: Separate models are built for each omics type, and their predictions are combined at the end. This is computationally efficient but may miss subtle interactions between different omics layers [12] [8].
4. How can I assess the success of a multi-omics data harmonization effort before moving to clinical validation? Success should be measured through a multi-tiered approach:
- Technical/Statistical Validation: Use metrics like the proportion of missing data resolved, reduction in batch effects, and the accuracy of imputation. Successful harmonization should also improve the performance of downstream models in classifying known sample groups (e.g., diseased vs. healthy) compared to non-harmonized data [2] [44].
- Biological Validation: The integrated data should recapitulate known biology and reveal novel, testable hypotheses. Pathway analysis on results from harmonized data should identify biologically plausible mechanisms [5] [2].
- Clinical Potential: The ultimate validation is the ability to identify robust biomarkers that can stratify patients, predict disease progression, or forecast drug response with higher accuracy than single-omics approaches [5] [83].
Troubleshooting Guides
Guide 1: Resolving Data Heterogeneity and Harmonization Failures
Problem: After combining datasets from different cohorts or labs, the data shows strong technical batch effects, and biological signals are obscured.
Investigation & Solution:
| Step | Action | Diagnostic Check |
|---|---|---|
| 1. Profile Data | Conduct a full inventory of all data sources. Assess data quality for missing values, inconsistent formats, and duplicate records [82]. | Use data profiling tools to generate reports on data types, value distributions, and outliers across all datasets [82]. |
| 2. Design Schema | Establish a common target schema and unified data model, such as the OMOP CDM in healthcare [82]. | Involve domain experts to ensure the schema reflects real-world needs and business logic for semantic accuracy [82]. |
| 3. Transform & Map | Execute syntactic and semantic mapping. Standardize formats (e.g., dates, units) and map different system codes to a single standard (e.g., map "M" and "1" to "Male") [82]. | Use ETL/ELT pipelines for automated transformation. Check that all data adheres to the predefined formats and value sets [82]. |
| 4. Validate | Run rigorous data quality checks to ensure the harmonized data conforms to the target schema and that known biological relationships are preserved [82]. | Programmatically verify data types and value constraints. Compare the output of a simple analysis (e.g., PCA) on harmonized vs. original data to check for reduced batch effects [12] [82]. |
Guide 2: Addressing Poor Model Performance Post-Integration
Problem: After integrating your omics data, your machine learning or statistical model shows poor performance, low predictive power, or an inability to find meaningful patterns.
Investigation & Solution:
| Symptom | Possible Cause | Solution |
|---|---|---|
| High dimensionality and overfitting. | The number of features (variables) is much larger than the number of samples (HDLSS problem) [8]. | Apply dimensionality reduction techniques (e.g., PCA, autoencoders) or use integration methods like MOFA that infer latent factors to reduce noise [12] [2]. |
| Inconsistent findings; model fails on new data. | Technical batch effects or non-biological variation were not adequately corrected during harmonization [12]. | Re-visit pre-processing. Apply batch effect correction algorithms (e.g., ComBat) and ensure proper experimental design to minimize these effects from the start [12]. |
| Model is complex but provides no biological insight. | The chosen integration method (e.g., early integration) created a "black box" [8]. | Switch to an interpretable method or one that provides factor loadings. Use DIABLO for supervised biomarker discovery or MOFA+ to identify latent factors that can be biologically annotated [2]. |
Experimental Protocols & Workflows
Detailed Protocol: A Multi-Omics Harmonization Pipeline for Cohort Integration
This protocol outlines a standardized workflow for harmonizing multi-omics data from disparate cohorts, as recommended by large-scale consortia like the NIH's Multi-Omics for Health and Disease (MOHD) and insights from recent literature [22] [83] [82].
1. Pre-Harmonization: Planning and Standardization
- Define Common Data Elements (CDEs): Before data collection, agree upon a minimal set of standardized variables (e.g., clinical phenotypes, sample processing metadata) to be collected uniformly across all sites [22].
- Adopt Metadata Standards: Use established metadata standards relevant to your data types (e.g., 3D Microscopy Metadata Standards for imaging, SPREC for biospecimens) to ensure all data is FAIR (Findable, Accessible, Interoperable, and Reusable) [22].
- Establish a Common Coordinate Framework (CCF): For spatial data, use a CCF to align high-resolution data into an appropriate anatomical context [22].
2. Data Processing and Harmonization Execution
- Step 1 - Raw Data Processing: Process raw data (e.g., FASTQ files for sequencing, raw spectra for proteomics) using standardized, version-controlled pipelines for each omics type to generate feature tables (e.g., gene counts, protein intensities) [22] [2].
- Step 2 - Normalization and Quality Control: Perform modality-specific normalization (e.g., TPM for RNA-seq, intensity-based for proteomics) and remove low-quality samples based on pre-defined QC metrics [12] [2].
- Step 3 - Batch Effect Correction: Identify and correct for batch effects using methods like ComBat or Harmony. Validate that batch effects are removed by visualizing data using PCA before and after correction [12].
- Step 4 - Semantic Harmonization: Map all identifiers (e.g., gene names, protein IDs, clinical terms) to controlled vocabularies or ontologies (e.g., HUGO Gene Nomenclature, SNOMED CT for clinical terms) to ensure semantic alignment [82].
3. Post-Harmonization Validation
- Check 1 - Technical Validation: Confirm that known control samples cluster together and that technical replicates are highly correlated in the harmonized dataset.
- Check 2 - Biological Validation: Verify that the harmonized data recapitulates known biological relationships (e.g., strong correlation between mRNA and protein levels for housekeeping genes).
Workflow Visualization: Multi-Omics Data Harmonization Pathway
The following diagram illustrates the logical flow of the harmonization process, from disparate data sources to an integrated, analysis-ready resource.
Methodologies: Key Multi-Omics Integration Algorithms
The table below summarizes the core algorithms and tools frequently used for integrating harmonized multi-omics datasets, as identified in recent reviews [2] [44].
| Method | Category | Brief Explanation | Primary Use Case |
|---|---|---|---|
| MOFA/MOFA+ [2] | Unsupervised, Factorization | A Bayesian framework that infers a set of latent factors that capture shared and specific sources of variation across multiple omics datasets. | Exploratory analysis of multi-omics data to identify major axes of variation without using sample labels. |
| DIABLO [2] | Supervised, Integration | Uses multiblock sPLS-DA to identify latent components that maximize separation between pre-defined sample groups and correlation between omics datasets. | Classification and biomarker discovery when sample groups (e.g., disease vs. control) are known. |
| SNF [2] [44] | Unsupervised, Network-based | Constructs sample-similarity networks for each omics type and then fuses them into a single network that captures shared information across all data types. | Clustering patients into molecular subtypes based on integrated multi-omics profiles. |
| WGCNA [44] | Unsupervised, Network-based | Identifies modules of highly correlated features (e.g., genes) within a single omics layer. Modules can then be correlated with other omics data or clinical traits. | Identifying co-expression networks and linking them to other biological layers or clinical outcomes. |
| xMWAS [44] | Correlation-based | Performs pairwise association analysis to build correlation networks between different omics datasets, identifying communities of interconnected features. | Uncovering associations between features from different omics layers (e.g., which metabolites correlate with which proteins). |
Visualization: Multi-Omics Integration Strategies
This diagram illustrates the three primary conceptual strategies for integrating multiple omics datasets, showing the stage at which data from different modalities are combined [12] [8].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key computational and data resources essential for conducting robust multi-omics harmonization and integration studies.
| Tool/Resource | Type | Function & Application |
|---|---|---|
| OMOP Common Data Model (CDM) [82] | Data Model | A standardized data model for observational health data, enabling the harmonization of electronic health records (EHRs) with omics data by providing a unified structure. |
| LOINC & SNOMED CT [82] | Ontology/Vocabulary | Controlled vocabularies for semantic harmonization. LOINC standardizes laboratory test codes, while SNOMED CT standardizes clinical terms, ensuring consistent meaning across datasets. |
| MOFA+ [2] | Software Package (R/Python) | A widely used tool for unsupervised integration of multi-omics data. It decomposes complex datasets into latent factors that represent shared and specific sources of variation. |
| MixOmics [2] | Software Package (R) | A comprehensive R toolkit that includes DIABLO for supervised multi-omics integration and other multivariate methods for dimension reduction and visualization. |
| ComBat [12] | Algorithm | A popular empirical Bayes method used to adjust for batch effects in high-dimensional data, helping to remove technical variation without erasing biological signals. |
| FAIR Principles [22] | Guidelines | A set of guiding principles (Findable, Accessible, Interoperable, Reusable) to ensure data is managed and curated in a way that enables maximal use and integration. |
Conclusion
Effective data harmonization is the cornerstone that unlocks the transformative potential of multi-omics studies, enabling a transition from isolated data points to a systems-level understanding of biology and disease. By adhering to FAIR principles, selecting appropriate integration methodologies, proactively addressing data quality issues, and rigorously validating findings, researchers can overcome the significant challenges of heterogeneity and scale. The future of biomedical research hinges on these practices, which will accelerate the development of personalized diagnostics and therapeutics, ultimately paving the way for a new era in precision medicine driven by robust, integrated biological insights.
References
- 1. 6 practices to begin with multi-omics data integration [https://www.pythiabio.com/post/6-practices-to-begin-with-multi-omics-data-integration]
- 2. Multi-Omics: Challenges and Solutions [https://bigomics.ch/blog/key-challenges-in-multi-omics-data-integration/]
- 3. Challenges in AI-driven multi-omics data analysis for ... [https://www.sciencedirect.com/science/article/pii/S2352914825000681]
- 4. Strategies for Comprehensive Multi-Omics Integrative Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592251/]
- 5. 2025 Trends: Multiomics [https://www.genengnews.com/topics/omics/2025-trends-multiomics/]
- 6. Harmonized Multi-Omics Data Analysis - How Does It ... [https://oxfordglobal.com/precision-medicine/resources/harmonized-multi-omics-data-analysis-how-does-it-impact-pharmaceutical-research]
- 7. A General Primer for Data Harmonization | Scientific Data [https://www.nature.com/articles/s41597-024-02956-3]
- 8. Challenges in multi-omics data integration [https://www.mindwalkai.com/blog/challenges-in-multi-omics-data-integration]
- 9. Data Standards for Omics Data: The Basis of Data Sharing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4152841/]
- 10. Best practices for clinical trials data harmonization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12083401/]
- 11. A guide to multi-omics data collection and integration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9747357/]
- 12. Integrating Multi-Omics for Precision Medicine: 2025 Guide [https://lifebit.ai/blog/integrating-multi-modal-genomic-and-multi-omics-data-for-precision-medicine/]
- 13. Data harmonization: Steps and best practices [https://datavid.com/blog/data-harmonization]
- 14. What Is Data Harmonization? Process, Challenges & Benefits [https://hevoacademy.com/data-quality/what-is-data-harmonization/]
- 15. Ten quick tips for avoiding pitfalls in multi-omics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10325053/]
- 16. A guide to multi-omics [https://frontlinegenomics.com/a-guide-to-multi-omics/]
- 17. FAIR Principles - GO FAIR [https://www.go-fair.org/fair-principles/]
- 18. What are the FAIR Data Principles? - Countway LibAnswers [https://asklib.hms.harvard.edu/nih-dmsp/faq/377100]
- 19. FAIR data principles: What you need to know [https://www.tiledb.com/blog/fair-data-principles-explained]
- 20. FAIR principles for Research - Data Management for Research [https://guides.library.cmu.edu/researchdatamanagement/FAIR_principles]
- 21. Data in the Evolving World of Life Sciences: Chaos to Order [https://greenleafgrp.com/insights/data-in-the-evolving-world-of-life-sciences-chaos-to-order/]
- 22. A Guide to Developing Harmonized Research Workflows in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12233188/]
- 23. Implementing FAIR Data Principles: A 2025 Researcher's ... [https://editverse.com/implementing-fair-data-principles-a-2025-researchers-guide/]
- 24. Metadata harmonization–Standards are the key for a better ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9233336/]
- 25. OHDSI Standardized Vocabularies-a large-scale ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/38175665/]
- 26. Metadata integrity in bioinformatics: Bridging the gap ... [https://www.sciencedirect.com/science/article/pii/S2001037023003616]
- 27. Integrating Multi-Omics and Medical Imaging in Artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12650882/]
- 28. A systematic review of intermediate fusion in multimodal ... [https://www.sciencedirect.com/science/article/pii/S0262885625000976]
- 29. A machine learning approach for multimodal data fusion ... [https://www.nature.com/articles/s41698-025-00917-6]
- 30. A Guide to Multi-omics Integration Strategies [https://frontlinegenomics.com/a-guide-to-multi-omics-integration-strategies/]
- 31. AI-driven multi-omics integration in precision oncology [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634751/]
- 32. Automated Data Harmonization in Clinical Research: Natural ... [https://formative.jmir.org/2025/1/e75608]
- 33. Guidance for Multi-Omics and Multi-Modal Data Integration ... [https://aws.amazon.com/solutions/guidance/multi-omics-and-multi-modal-data-integration-and-analysis/]
- 34. Mastering Data Preprocessing: A Practical Guide to Clean ... [https://medium.com/@shrutikakkapade21/mastering-data-preprocessing-a-practical-guide-to-clean-and-transform-data-for-machine-learning-74d151dd523e]
- 35. A comparative study of imputation techniques for missing ... [https://link.springer.com/article/10.1007/s41060-025-00825-9]
- 36. Missing Data Imputation Techniques [https://www.nature.com/research-intelligence/nri-topic-summaries/missing-data-imputation-techniques-micro-41849]
- 37. Stop Wasting Time: Correcting Batch Effects in Multi-Omics ... [https://pluto.bio/resources/Learning%20Series/batch-effects-multi-omics-data-analysis]
- 38. High performance data integration for large-scale analyses ... [https://www.nature.com/articles/s41467-025-62237-4]
- 39. What is Data Normalization? A Complete Guide for 2025 [https://improvado.io/blog/data-normalization]
- 40. How to Normalize Data (2025 Guide) - Knack [https://www.knack.com/blog/how-to-normalize-data/]
- 41. Evaluation of normalization strategies for mass spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12214035/]
- 42. Chapter 2 — Designing a Multiomics Study [https://www.metabolon.com/multiomics-guide/designing-a-multiomics-study/]
- 43. Answering Your Questions about Sample Prep ... [https://www.10xgenomics.com/blog/answering-your-questions-about-sample-prep-optimization-for-single-cell-multiomics]
- 44. Algorithms and tools for data-driven omics integration to ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06446-x]
- 45. Requirements for Troubleshooting [https://omicsforum.ca/t/requirements-for-troubleshooting/857]
- 46. Guide to Creating an Analtyics Federated Operating Model ... [https://iianalytics.com/federated-analytics]
- 47. The Complete Guide to Federated Data Analysis [https://lifebit.ai/blog/federated-data-analysis-2/]
- 48. OmicsIntegrator - Systems Biology of Disease - Fraenkel Lab [https://fraenkel-nsf.csbi.mit.edu/omicsintegrator/]
- 49. 14 Most Common Data Quality Issues and How to Fix Them [https://lakefs.io/data-quality/data-quality-issues/]
- 50. 10 Common Data Quality Issues (And How to Solve Them) [https://firsteigen.com/blog/10-common-data-quality-issues-and-how-to-solve-them/]
- 51. 12 Best Data Quality Tools for 2025 [https://lakefs.io/data-quality/data-quality-tools/]
- 52. The 10 Best Data Quality Assessment Tools Of August 2025 [https://www.montecarlodata.com/blog-best-quality-assessment-tools/]
- 53. Data Quality Assurance at Research Data Repositories [https://datascience.codata.org/articles/dsj-2022-018]
- 54. Dealing with dimensionality: the application of machine learning to multi-omics data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9907220/]
- 55. From Silos to Synthesis: A comprehensive review of ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525004597]
- 56. Systems Biology and Multi-Omics Integration - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6523452/]
- 57. A practical guide to FAIR data management in the age of multi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11788310/]
- 58. An Introduction to Systems Analytics and Integration of Big ... [https://www.mdpi.com/2073-4425/11/3/245]
- 59. Data Pipelines: Key Components and Best Practices [https://www.getdbt.com/blog/data-pipelines]
- 60. Data pipeline best practices for cost optimization and ... [https://xenoss.io/blog/data-pipeline-best-practices]
- 61. 7 Best Practices for Designing Automated Data Pipelines in ... [https://www.shakudo.io/blog/7-best-practices-for-designing-automated-data-pipelines]
- 62. Dataflow pipeline best practices [https://docs.cloud.google.com/dataflow/docs/guides/pipeline-best-practices]
- 63. Federated data sharing: Your Ultimate 2025 Guide [https://lifebit.ai/blog/federated-data-sharing-complete-guide/]
- 64. PPML-Omics: A privacy-preserving federated machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10830108/]
- 65. A scoping review of the governance of federated learning in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12246253/]
- 66. Data governance framework: What it is, best practices & ... [https://www.fivetran.com/learn/data-governance-framework]
- 67. A Privacy-Preserving Federated Learning Method with ... [https://arxiv.org/html/2511.06064v1]
- 68. Data Privacy Trends Shaping 2025 and the Years Ahead [https://usercentrics.com/guides/data-privacy/data-privacy-trends/]
- 69. Privacy + Data Security Predictions for 2025 [https://www.mofo.com/resources/insights/250107-privacy-data-security-predictions]
- 70. Multitask benchmarking of single-cell multimodal omics ... [https://www.nature.com/articles/s41592-025-02856-3]
- 71. AI and the future of biomarker analysis in early R&D [https://www.drugtargetreview.com/article/190428/ai-and-the-future-of-biomarker-analysis-in-early-rd/]
- 72. Biomarker discovery studies for patient stratification using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8650485/]
- 73. Patient stratification biomarkers [https://precisionlife.com/biomarkers]
- 74. Introduction to multi-omics data integration and visualisation [https://www.ebi.ac.uk/training/events/introduction-multi-omics-data-integration-and-visualisation-2025]
- 75. Navigating Cancer Complexity: Integrative Multi-Omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553891/]
- 76. Scrutinizing neurodegenerative diseases: decoding the ... [https://humgenomics.biomedcentral.com/articles/10.1186/s40246-024-00704-7]
- 77. Why Multi-Omics Data Integration Often Fails [https://www4.accurascience.com/blogs_44_0.html]
- 78. Flexynesis: A deep learning toolkit for bulk multi-omics data ... [https://www.nature.com/articles/s41467-025-63688-5]
- 79. A review on multi-omics integration for aiding study design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12372284/]
- 80. Multi-omics data integration using ratio-based quantitative ... [https://www.nature.com/articles/s41587-023-01934-1]
- 81. Data Harmonization Techniques: 5 Steps to Seamless [https://lifebit.ai/blog/data-harmonization-techniques/]
- 82. Data Harmonization Meaning: 5 Steps to Flawless Harmony [https://lifebit.ai/blog/data-harmonization-meaning-complete-guide/]
- 83. Multi-Omics for Health and Disease (MOHD) [https://www.genome.gov/research-funding/Funded-Programs-Projects/Multi-Omics-for-Health-and-Disease]