Breaking the Barrier: Innovative Strategies to Overcome Intrinsic Resistance in Gram-Negative Bacteria
The unique structure of the Gram-negative cell envelope, particularly the asymmetric outer membrane, confers formidable intrinsic resistance to a wide range of antibiotics, making infections caused by pathogens like Acinetobacter...
Breaking the Barrier: Innovative Strategies to Overcome Intrinsic Resistance in Gram-Negative Bacteria
Abstract
The unique structure of the Gram-negative cell envelope, particularly the asymmetric outer membrane, confers formidable intrinsic resistance to a wide range of antibiotics, making infections caused by pathogens like Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae a critical global health threat. This article provides a comprehensive resource for researchers and drug development professionals, exploring the foundational science behind intrinsic resistance mechanisms, evaluating current and emerging methodological approaches to circumvent these defenses—including antibiotic adjuvants, antimicrobial peptides (AMPs), and nanocarrier systems—and discussing the optimization and validation of these novel therapies. By synthesizing insights from foundational exploration to comparative analysis of pipeline candidates, this review aims to guide the strategic development of next-generation antimicrobials capable of overcoming one of the most pressing challenges in modern medicine.
Deconstructing the Fortress: The Gram-Negative Cell Envelope and Innate Defense Mechanisms
Frequently Asked Questions (FAQs)
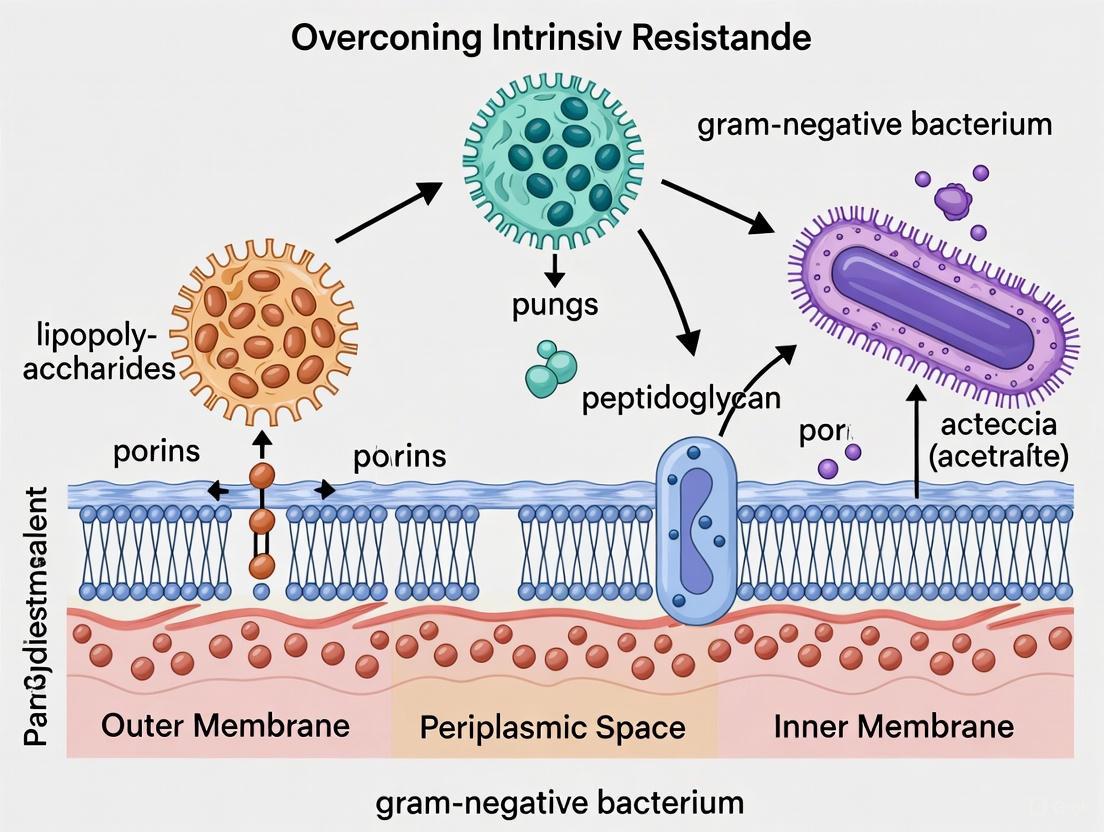
1. What makes the outer membrane of Gram-negative bacteria asymmetric, and why is this significant for antibiotic resistance? The outer membrane is asymmetric because its inner leaflet is composed of phospholipids (like phosphatidylethanolamine, phosphatidylglycerol, and cardiolipin), while the outer leaflet is built from glycolipids, primarily lipopolysaccharides (LPS) [1] [2]. This unique architecture creates a formidable permeability barrier. The dense, negatively charged LPS layer, stabilized by divalent cations, strongly inhibits the penetration of many hydrophobic antibiotics, bile salts, and detergents, conferring intrinsic resistance to Gram-negative bacteria [2] [3].
2. How can I experimentally reconstitute an asymmetric model membrane to study its properties? You can use several well-established techniques to create asymmetric membranes that mimic the bacterial outer membrane [1]:
- Montal-Mueller Technique: Forms planar lipid bilayers across a small aperture, ideal for electrical measurements of channel activity.
- Langmuir-Blodgett Technique: Creates solid-supported monolayers or bilayers by sequentially transferring lipid layers onto a solid substrate.
- Phase Transfer Method: Used to prepare asymmetric Giant Unilamellar Vesicles (aGUVs), allowing for visualization of phase behavior and domain formation under a microscope. A study successfully created aGUVs with LPS R45 on one leaflet and a phospholipid mixture on the other [1].
3. My antimicrobial agent is ineffective in vivo despite showing promise in vitro. Could the asymmetric membrane be a factor? Yes, absolutely. The asymmetric LPS-phospholipid structure is a key reason for the discrepancy between drug efficacy in laboratory tests (in vitro) and in living organisms (in vivo). In vitro assays often use simplified, symmetric membranes or bacterial strains with compromised outer membranes. The native, asymmetric membrane significantly reduces the uptake of many compounds. Your experiments should include robust asymmetric model membranes or genetically intact bacterial strains to better predict clinical outcomes [1] [3].
4. What are the primary functions of porins, and how do bacteria modify them to resist antibiotics? Porins are beta-barrel proteins in the outer membrane that form water-filled channels for the passive diffusion of small, hydrophilic molecules [4] [5] [6]. They act as a molecular sieve, typically with an exclusion limit of around 600 Da [6]. Bacteria develop resistance through porin modifications such as:
- Downregulation: Reducing the number of porin channels to limit antibiotic entry.
- Mutation: Altering the amino acids lining the channel to change its size or charge, thereby hindering the passage of specific antibiotics like β-lactams and fluoroquinolones [3] [5].
5. What is the difference between "smooth" and "rough" LPS, and how does this impact membrane permeability?
- Smooth LPS: Contains the full O-antigen polysaccharide chain, creating a dense, protective surface that is less permeable to hydrophobic molecules.
- Rough LPS: Lacks the O-antigen or has a truncated core oligosaccharide. This "rough" form creates a more hydrophobic and penetrable membrane, making the bacteria more susceptible to some hydrophobic antibiotics [1] [7].
Troubleshooting Guides
Issue 1: Low Antibiotic Permeability in Asymmetric Membrane Models
Problem: Your experimental data shows unexpectedly low permeability of an antibiotic compound through your asymmetric membrane model. Potential Causes and Solutions:
- Cause: Incorrect LPS Leaflet Density. The outer LPS leaflet may not be packed densely enough, failing to replicate the native barrier.
- Solution: For Langmuir-Blodgett films, ensure precise control of the surface pressure during monolayer transfer. For aGUVs, verify the phase transfer protocol and the purity of the LPS stock [1].
- Cause: Porin Malfunction or Absence. If your model incorporates porins, they may not be correctly folded or inserted into the membrane.
- Cause: Cation Depletion. The integrity of the LPS layer is stabilized by divalent cations (e.g., Mg²⁺).
Issue 2: Inconsistent Results with LPS-Dependent Assays
Problem: High background noise or inconsistent data in assays measuring interactions with LPS (e.g., binding or immune activation). Potential Causes and Solutions:
- Cause: LPS Contamination.
- Solution: Use LPS-free reagents, tubes, and tips. Validate the absence of contaminating LPS in your buffer systems using a Limulus Amebocyte Lysate (LAL) test.
- Cause: Variable LPS Aggregation State.
- Solution: Standardize the preparation of LPS suspensions. Use a consistent sonication or heating protocol to create uniform micelles or vesicles before each experiment [7].
- Cause: Use of Non-Physiological LPS Forms.
Issue 3: Porin Expression and Channel Activity Loss in Mutant Strains
Problem: Your bacterial mutant, created to study a specific porin, shows no channel activity or unexpected physiological defects. Potential Causes and Solutions:
- Cause: Impropor Outer Membrane Assembly.
- Cause: Compensatory Mutations.
- Solution: Bacteria may upregulate other porins or efflux pumps to compensate for the loss of a specific porin. Whole-genome sequence your mutant to identify secondary mutations and characterize the expression levels of other major outer membrane proteins [3].
- Cause: Protein Misfolding.
- Solution: Verify that the porin is correctly targeted to the outer membrane and not accumulating in the periplasm. Use antibodies to check for the presence of the protein and its trimeric, mature form [4].
Experimental Data & Protocols
Quantitative Data on Membrane Properties
Table 1: Lipid Composition of Model Asymmetric Membranes for Gram-Negative Bacteria Research [1]
| Membrane Leaflet | Lipid Components | Example Ratio (Salmonella typhimurium) | Function in Model |
|---|---|---|---|
| Outer (External) | Lipopolysaccharide (LPS) | 100% | Primary permeability barrier; endotoxin activity |
| Inner (Periplasmic) | Phosphatidylethanolamine (PE) | 81% | Main structural phospholipid |
| Phosphatidylglycerol (PG) | 17% | Contributes to membrane charge | |
| Cardiolipin (DPG) | 2% | Found in membrane domains, involved in stress response |
Table 2: Common Porins and Their Characteristics in Gram-Negative Bacteria [4] [5] [6]
| Porin Name | Organism | Channel Properties | Exclusion Limit (Approx.) | Key Features |
|---|---|---|---|---|
| OmpF | E. coli | General diffusion, slightly cation-selective | ~600 Da | Major porin; trimeric structure; expression regulated by osmolarity |
| OmpC | E. coli | General diffusion, smaller than OmpF | ~600 Da | Major porin; expressed at high osmolarity |
| PhoE | E. coli | General diffusion, anion-selective | ~600 Da | Induced under phosphate starvation |
| OprP | P. aeruginosa | Specific channel | N/A | Highly specific for phosphate |
| Tsx | E. coli | Specific channel | N/A | Specific for nucleosides |
Detailed Experimental Protocol: Phase Transfer Method for Asymmetric GUVs
This protocol is adapted from Pautot et al. (2003) and subsequent work for creating aGUVs with an LPS outer leaflet and a phospholipid inner leaflet [1].
Objective: To generate giant unilamellar vesicles (GUVs) with asymmetric lipid distribution for biophysical studies, such as phase behavior analysis or peptide interaction assays.
Materials:
- Lipids: Purified LPS (e.g., R45 from Proteus mirabilis), PE, PG, CL.
- Solvents: Chloroform, ethanol, methanol.
- Oils: Pure anhydrous dodecane, silicone oil (AR 200).
- Buffers: 100 mM KCl, 5 mM MgCl₂, 5 mM HEPES, pH 7.0.
- Labware: Glass vials, Hamilton syringes, phase-transfer tube.
Method:
- Forming the Lipid-Oil Solution:
- Dissolve the inner leaflet lipids (PL mixture: PE/PG/CL at 81:17:2) in a mixture of dodecane and silicone oil (99:1 v/v) to a final concentration of 0.5-1 mM.
- In a separate vial, dissolve the outer leaflet lipid (LPS) in an organic solvent (e.g., chloroform/methanol). Then, add this solution to pure dodecane and evaporate the organic solvent under a nitrogen stream to obtain LPS dissolved in dodecane.
Lipid Monolayer Formation:
- Add an aliquot of the inner leaflet lipid-oil solution on top of the aqueous buffer in a phase-transfer tube. A lipid monolayer will form at the oil-water interface.
Phase Transfer and Vesicle Formation:
- Carefully layer a small volume of the outer leaflet lipid-oil (LPS) solution on top of a dense sucrose cushion in a centrifuge tube.
- Gently transfer the tube containing the inner leaflet monolayer through the outer leaflet solution. As it passes through, the outer leaflet lipids assemble on the droplet.
- Centrifuge the tube to pull the now-coated droplets through the sucrose cushion into the underlying buffer solution. This process forms the aGUVs.
Harvesting and Characterization:
- Collect the aGUVs from the bottom of the tube.
- Characterize asymmetry using fluorescence techniques, for example, by incorporating leaflet-specific fluorescent lipids and performing dithionite quenching assays [1].
Signaling Pathways and Experimental Workflows
Membrane Asymmetry and Resistance Pathways
Diagram: Membrane Structure Role in Resistance
Experimental Workflow for Porin Function
Diagram: Porin Functional Analysis Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Studying the Gram-Negative Outer Membrane
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| Deep Rough Mutant LPS | Creating model membranes with a defined, "leaky" LPS layer for permeability studies. | LPS R45 from Proteus mirabilis; lacks O-antigen for easier handling [1]. |
| Smooth-Form LPS | Studying immune activation (e.g., TLR4 signaling) and the full native barrier function. | Commercially available from E. coli or Salmonella; contains full O-antigen chain [2] [7]. |
| Purified Porins | Functional reconstitution experiments to study solute diffusion and channel gating. | OmpF, OmpC from E. coli; used in liposome swelling or planar bilayer assays [4] [6]. |
| Polymyxin B Nonapeptide (PMBN) | A benchmark outer membrane permeabilizer with reduced toxicity; used as an adjuvant in synergy studies [3]. | |
| Divalent Cations | Critical for stabilizing the LPS layer by bridging negative charges; essential in buffers. | MgCl₂, CaCl₂; typically used at 1-5 mM concentration [1] [2]. |
| Acyloxyacyl Hydrolase (AOAH) | Enzyme used to detoxify LPS; research tool for studying LPS-induced immune signaling [7]. | Inactivates LPS by removing secondary acyl chains from lipid A. |
Frequently Asked Questions (FAQs)
Q1: What are the primary mechanisms that constitute the "Multifaceted Shield" of intrinsic resistance in Gram-negative bacteria? The intrinsic resistance of Gram-negative bacteria is primarily built upon three core mechanisms that function synergistically [8] [9]:
- Limited Uptake: The asymmetric outer membrane, featuring lipopolysaccharide (LPS) and restrictive porin channels, acts as a formidable barrier to many antibiotics, particularly hydrophobic compounds and large molecules [8] [10].
- Efflux Pumps: Energy-dependent transporters, especially those from the Resistance Nodulation Division (RND) superfamily, actively pump a wide range of antibiotics out of the cell, reducing intracellular concentrations [11] [12].
- Drug Inactivation: Enzymes located in the periplasm or cytoplasm can modify or degrade antibiotics before they reach their target [8] [13]. These mechanisms are innate to the bacterial species and are independent of prior antibiotic exposure or horizontal gene transfer [14] [9].
Q2: Why are Gram-negative bacteria intrinsically resistant to many antibiotics that are effective against Gram-positive bacteria? The key differentiator is the complex cell envelope of Gram-negative bacteria [8] [3]. Unlike Gram-positive bacteria, which have a single cytoplasmic membrane and a thick peptidoglycan layer, Gram-negative bacteria possess an additional outer membrane. This outer membrane is asymmetric, with a dense layer of LPS in the outer leaflet that impedes the penetration of hydrophobic molecules [10] [15]. Furthermore, the entry of hydrophilic molecules is limited to porin channels, which restrict the size and type of compounds that can diffuse through [10] [3]. This physical barrier, combined with potent efflux pumps, creates a powerful defensive shield [14].
Q3: My experimental data shows a high Minimum Inhibitory Concentration (MIC) for a novel compound against a Gram-negative pathogen. How can I determine if efflux pumps are responsible? A significant increase in MIC in the presence of an efflux pump inhibitor (EPI) is a strong indicator of efflux involvement. Below is a standardized protocol to investigate this.
Table: Experimental Protocol for Efflux Pump Inhibition Assay
| Step | Action | Purpose & Notes |
|---|---|---|
| 1. Bacterial Strain | Use your clinical/test isolate and a control strain (e.g., E. coli ATCC 25922). | Provides a baseline for comparison. |
| 2. Efflux Pump Inhibitor (EPI) | Prepare a sub-inhibitory concentration of an EPI like PaβN (Phe-Arg-β-naphthylamide). Common working concentration: 20-50 µg/mL. | Inhibits RND-family pumps; a sub-inhibitory concentration avoids killing the bacteria. |
| 3. Broth Microdilution | Perform MIC assays in duplicate: (a) Antibiotic/compound alone, (b) Antibiotic/compound + EPI. | The gold-standard method for susceptibility testing. |
| 4. Interpretation | A ≥4-fold decrease in MIC in the presence of the EPI is considered a positive result for efflux involvement. | Indicates the pump is actively extruding your compound. |
Q4: In a susceptibility test, my bacterial isolate is resistant to a β-lactam/β-lactamase inhibitor combination, but no common β-lactamase genes are detected. What other mechanisms should I investigate? This scenario points towards non-enzymatic mechanisms. Your investigation should focus on:
- Porin Mutations: Sequence the major porin genes (e.g., OmpF, OmpC in E. coli; OprD in P. aeruginosa). Mutations leading to loss or reduction of these porins can significantly limit the influx of β-lactams, even when β-lactamases are inhibited [12] [3].
- Hyperactive Efflux Pumps: Investigate the expression levels of RND efflux pumps (e.g., AcrAB-TolC in E. coli; MexAB-OprM in P. aeruginosa). Mutations in regulatory genes (e.g., marA, soxS, ramA) can lead to pump overexpression, enabling the extrusion of β-lactams and novel combinations like ceftazidime/avibactam [12].
- Target Site Modification: Although less common, alterations in penicillin-binding proteins (PBPs) can reduce the binding affinity of the β-lactam antibiotic [12].
Q5: What are the most promising therapeutic strategies being developed to breach this intrinsic resistance? Current research is focused on two main adjuvant strategies to potentiate existing antibiotics [10] [3] [15]:
- Membrane Permeabilizers: Compounds like polymyxin B nonapeptide (PMBN) and its newer derivatives (e.g., SPR741) disrupt the integrity of the outer membrane, allowing otherwise excluded antibiotics to enter the cell [3] [15].
- Efflux Pump Inhibitors (EPIs): The development of small molecules that block the function of RND efflux pumps. When co-administered with an antibiotic, EPIs can restore the antibiotic's activity by preventing its extrusion [11] [3]. These approaches aim to "re-sensitize" Gram-negative pathogens to a broader spectrum of antibiotics.
Troubleshooting Common Experimental Challenges
Table: Troubleshooting Guide for Intrinsic Resistance Research
| Problem | Potential Cause | Recommended Solution | Supporting Experimental Approach |
|---|---|---|---|
| High MIC for a new compound against a Gram-negative panel. | The compound is a substrate for broad-spectrum efflux pumps. | Co-administer with an efflux pump inhibitor (EPI). | Perform a broth microdilution MIC assay with and without a known EPI like PaβN [11]. |
| Inconsistent results in membrane permeabilization assays. | Unstable or degraded permeabilizing agent; incorrect sub-inhibitory concentration. | Prepare fresh stocks of the adjuvant and confirm its sub-inhibitory concentration for each strain. | Use a positive control like polymyxin B nonapeptide and validate its activity in a standalone MIC assay [15]. |
| Suspected porin-mediated resistance, but sequencing reveals no mutations. | Down-regulation of porin gene expression. | The resistance is likely transcriptional, not mutational. | Quantify porin gene expression using RT-qPCR, comparing the isolate to a wild-type control strain [3]. |
| An engineered compound shows good in vitro activity but fails in an animal model. | In vivo efflux or sequestration of the compound; toxicity issues. | Re-evaluate the compound's pharmacokinetic and toxicity profile. | Test for synergy with an EPI in the in vivo model and conduct thorough toxicological studies [11] [10]. |
| Difficulty in cloning or expressing efflux pump genes. | Toxicity of the pump gene to the expression host. | Use a tightly regulated, inducible expression vector and optimize induction conditions. | Clone the gene into a vector with an inducible promoter (e.g., pET with T7/lac) and titrate the inducer (e.g., IPTG) [11]. |
The Scientist's Toolkit: Essential Research Reagents
Table: Key Reagents for Studying Intrinsic Resistance Mechanisms
| Reagent / Material | Primary Function in Research | Example Application |
|---|---|---|
| Phe-Arg-β-naphthylamide (PaβN) | A broad-spectrum efflux pump inhibitor (EPI) targeting RND family pumps. | Used in MIC assays to confirm and characterize efflux-mediated resistance [11]. |
| Polymyxin B Nonapeptide (PMBN) | An outer membrane permeabilizer that lacks direct antibacterial activity. | Used as a positive control in synergy studies to sensitize bacteria to large or hydrophobic antibiotics [3] [15]. |
| Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) | A proton motive force (PMF) uncoupler. | Used to distinguish between PMF-dependent efflux (e.g., RND, MFS) and ATP-dependent efflux (e.g., ABC transporters) [11]. |
| Ethidium Bromide | A fluorescent efflux pump substrate. | Used in real-time fluorometric assays (e.g., using a spectrophotometer) to visualize and quantify efflux pump activity. |
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | The standardized medium for antibiotic susceptibility testing. | Essential for performing reproducible and clinically relevant broth microdilution MIC assays. |
| Isogenic Mutant Strains | Engineered strains with specific gene deletions (e.g., ΔacrB, ΔtolC). | Used as negative controls to definitively link a specific efflux pump to resistance against a compound of interest [11] [12]. |
Visualizing Core Concepts and Workflows
The Tripartite RND Efflux Pump Complex
Diagram 1: Antibiotic Extrusion via RND Efflux Pump
Experimental Workflow for Resistance Mechanism Investigation
Diagram 2: Systematic Investigation of Intrinsic Resistance
Standard Experimental Protocols
Protocol: Checkerboard Synergy Assay for Adjuvant Screening
Purpose: To quantitatively determine the synergistic effect between an antibiotic and a potential adjuvant (e.g., EPI or membrane permeabilizer). Materials:
- Cation-adjusted Mueller-Hinton Broth (CAMHB)
- Sterile 96-well microtiter plates
- Bacterial suspension adjusted to 0.5 McFarland standard
- Antibiotic stock solution
- Adjuvant stock solution
Procedure:
- Plate Preparation: Prepare a 2D dilution series. Serially dilute the antibiotic along the x-axis (e.g., columns 1-12) and the adjuvant along the y-axis (e.g., rows A-H).
- Inoculation: Dilute the bacterial suspension in CAMHB and add to each well, achieving a final inoculum of ~5 × 10^5 CFU/mL. Leave column 12 and row H as sterile controls.
- Incubation: Incubate the plate at 35°C for 16-20 hours.
- Calculation & Interpretation: Calculate the Fractional Inhibitory Concentration Index (FICI).
Protocol: Real-Time Ethidium Bromide Efflux Assay
Purpose: To visually and quantitatively assess the activity of efflux pumps in live bacterial cells. Principle: Ethidium bromide (EtBr) fluoresces intensely when bound to DNA inside the cell. Active efflux pumps extrude EtBr, reducing fluorescence. Inhibition of pumps leads to intracellular accumulation and increased fluorescence. Materials:
- Bacterial culture in mid-log phase
- Ethidium Bromide stock solution
- Efflux Pump Inhibitor (e.g., PaβN)
- Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) - positive control
- Phosphate Buffered Saline (PBS) or HEPES buffer
- Spectrofluorometer or fluorescence microplate reader
Procedure:
- Cell Loading: Wash and resuspend bacterial cells in buffer. Load the cells with a sub-inhibitory concentration of EtBr in the presence of the energy poison CCCP (e.g., 50 µM) for 30-60 minutes to allow accumulation without active efflux.
- Washing: Centrifuge the cells and wash twice with warm buffer to remove extracellular EtBr and CCCP.
- Efflux Measurement: Resuspend the cells in buffer and immediately dispense into a 96-well plate. Add the test EPI to the experimental wells. Monitor fluorescence over time (e.g., every 5-10 minutes for 60 minutes) with excitation at 530 nm and emission at 600 nm.
- Interpretation: Strains with high efflux activity will show a rapid decrease in fluorescence. A significant increase in fluorescence retention in the presence of an EPI indicates successful inhibition of the efflux pump [11].
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) represent a group of nosocomial pathogens that exhibit multidrug resistance and virulence, enabling them to "escape" the biocidal action of antimicrobial agents [16] [17]. Among these, A. baumannii, P. aeruginosa, and K. pneumoniae are classified as Critical Priority by the World Health Organization (WHO) due to their extensive drug resistance profiles and significant mortality rates [16] [18]. These Gram-negative bacteria are responsible for life-threatening infections including ventilator-associated pneumonia, bloodstream infections, urinary tract infections, and surgical site infections, particularly affecting critically ill and immunocompromised patients [19] [17].
The global burden of antimicrobial resistance (AMR) is substantial, resulting in an estimated 4.95 million deaths annually [19]. A 2025 study in southern Ethiopia found a shocking 76.8% prevalence of culture-confirmed surgical site infections among adult patients who underwent major surgery, with ESKAPE pathogens comprising 65.3% of the isolates [19]. The same study revealed that 84.37% of ESKAPE pathogens exhibited multidrug resistance (MDR), with A. baumannii showing the highest MDR rate at 100%, followed by K. pneumoniae at 88.23% [19]. The COVID-19 pandemic has exacerbated this pre-existing crisis, with one report noting that 68.9% of COVID-19 patients used antibiotics before hospitalization, accelerating the development of resistance [18].
Resistance Mechanisms in WHO-Critical Gram-Negative Pathogens
Understanding the complex resistance mechanisms of these pathogens is crucial for developing effective countermeasures. These bacteria employ a multifaceted arsenal of defense strategies, encompassing intrinsic, adaptive, and acquired resistance mechanisms.
Intrinsic Resistance Mechanisms
Pseudomonas aeruginosa presents one of the most formidable intrinsic resistance profiles among clinically relevant bacteria, limiting available treatment options even for wild-type isolates [20]. This intrinsic resistance is mediated through:
- Reduced Outer Membrane Permeability: The hydrophobic outer membrane bilayer, containing lipopolysaccharide (LPS) molecules, acts as a barrier to many antibiotics [21]. Hydrophilic antibiotics like β-lactams must use channel proteins (porins) for entry, while hydrophobic antibiotics diffuse through lipids [21].
- Chromosomally Encoded Antibiotic-Inactivating Enzymes: This includes inducible AmpC cephalosporinases and other β-lactamases that hydrolyze penicillins and cephalosporins [20].
- Efflux Pump Systems: P. aeruginosa utilizes several Resistance-Nodulation-Division (RND) efflux pumps including MexAB-OprM, MexXY-OprM, MexCD-OprJ, and MexEF-OprN that expel a wide array of antimicrobials including fluoroquinolones, β-lactams, macrolides, tetracyclines, and aminoglycosides [20].
Acinetobacter baumannii and Klebsiella pneumoniae share similar intrinsic defense strategies, though with some variation in their specific mechanisms and efficiency.
Acquired Resistance Mechanisms
The acquisition of mobile genetic elements through horizontal gene transfer significantly amplifies the threat posed by these pathogens. The primary acquired resistance mechanisms include:
- Extended-Spectrum β-Lactamase (ESBL) Production: Particularly in K. pneumoniae, with common enzymes including TEM, SHV, and CTX-M types that confer resistance to penicillins, cephalosporins, and aztreonam [17].
- Carbapenemase Production: These enzymes hydrolyze carbapenems, last-resort antibiotics for MDR infections. Key enzymes include:
- Aminoglycoside-Modifying Enzymes: Conferring resistance to aminoglycoside antibiotics [20].
- Modification of Drug Targets: Mutations in DNA gyrase and topoisomerase IV leading to fluoroquinolone resistance [20].
- Overexpression of Efflux Pumps: Increased expression of intrinsic efflux systems through mutation [20].
Adaptive Resistance: The Biofilm Challenge
All three pathogens demonstrate significant adaptive resistance through biofilm formation. Biofilms are structured communities of bacterial cells enclosed in an extracellular polymeric matrix that physically restricts antibiotic penetration and creates heterogeneous microenvironments with specialized, dormant persister cells that exhibit extreme antibiotic tolerance [16]. This makes biofilm-associated infections particularly challenging to eradicate, contributing to chronic infections in medical devices and compromised tissues.
Table 1: Major Resistance Mechanisms in WHO-Critical ESKAPE Pathogens
| Pathogen | Intrinsic Mechanisms | Key Acquired Resistance Enzymes | Efflux Systems |
|---|---|---|---|
| A. baumannii | Limited outer membrane permeability, Chromosomal OXA-51 | OXA-type carbapenemases (OXA-23, OXA-58), MBLs (NDM, VIM, IMP) | AdeABC, AdeFGH |
| P. aeruginosa | AmpC cephalosporinase, Low outer membrane permeability, Efflux pumps | ESBLs (PER, VEB), MBLs (IMP, VIM), KPC (rare) | MexAB-OprM, MexXY-OprM |
| K. pneumoniae | Capsular polysaccharide barrier | ESBLs (CTX-M, TEM, SHV), KPC carbapenemases, MBLs (NDM, VIM) | AcrAB-TolC |
Table 2: Documented Resistance Rates in Clinical Settings (2020-2025)
| Pathogen | ESBL Production | Carbapenem Resistance | MDR Rate | Key References |
|---|---|---|---|---|
| A. baumannii | Not routinely tested | >80% (Global, 2022) | 100% (Ethiopian study, 2025) | [19] [18] |
| P. aeruginosa | Not routinely tested | ~25% (Global, 2022) | 84.37% (ESKAPE collective, 2025) | [19] [18] |
| K. pneumoniae | 33.9% (Ethiopian study, 2025) | Increasing globally | 88.23% (Ethiopian study, 2025) | [19] |
Experimental Workflows for Resistance Profiling
Standard Antimicrobial Susceptibility Testing (AST)
Purpose: To determine the Minimum Inhibitory Concentration (MIC) of antibiotics against clinical isolates and classify them as Susceptible, Intermediate, or Resistant based on established clinical breakpoints.
Methodology (Kirby-Bauer Disk Diffusion) [19]:
- Bacterial Inoculum Preparation: Adjust the turbidity of a bacterial suspension in saline or broth to match the 0.5 McFarland standard (approximately 1.5 × 10^8 CFU/mL).
- Inoculation: Evenly spread the standardized inoculum over the surface of Mueller-Hinton agar plates using a sterile cotton swab.
- Antibiotic Disk Application: Aseptically place antibiotic-containing disks on the inoculated agar surface using sterile forceps, ensuring adequate spacing (center-to-center distance of 24 mm) to prevent inhibition zone overlap.
- Incubation: Incubate plates at 35°C ± 2°C for 16-18 hours in ambient air.
- Measurement and Interpretation: Measure the diameter of inhibition zones including the disk diameter using calipers or a ruler. Interpret results according to Clinical and Laboratory Standards Institute (CLSI) guidelines.
Key Reagents:
- Mueller-Hinton Agar
- Antibiotic disks
- McFarland standards
- Sterile saline
Phenotypic Detection of Specific Resistance Mechanisms
Extended-Spectrum β-Lactamase (ESBL) Detection - Combined Disk Method [19]:
- Inoculum Preparation: Standardize bacterial suspension to 0.5 McFarland.
- Disk Application: Place disks containing cefotaxime (30 µg) and ceftazidime (30 µg) alone and in combination with clavulanic acid (cefotaxime/clavulanic acid and ceftazidime/clavulanic acid) on the inoculated agar.
- Incubation and Interpretation: Incubate at 35°C for 16-18 hours. An increase of ≥5 mm in the inhibition zone diameter for either antimicrobial agent tested in combination with clavulanic acid versus its zone when tested alone confirms ESBL production.
Carbapenemase Detection - Modified Carbapenem Inactivation Method (mCIM):
- Inoculum Preparation: Create a 1 µL loop of test isolate emulsified in 2 mL of Tryptic Soy Broth.
- Incubation with Antibiotic: Add a 10 µg meropenem disk to the broth and incubate at 35°C for 4 hours.
- Indicator Lawn: Prepare a 0.5 McFarland suspension of E. coli ATCC 25922 and lawn on Mueller-Hinton agar.
- Disk Transfer: Remove the meropenem disk from the broth and place on the inoculated agar plate.
- Final Incubation and Interpretation: Incubate at 35°C for 18-24 hours. A zone diameter of 6-15 mm or presence of colonies within a 16-18 mm zone indicates a positive result for carbapenemase production.
Laboratory Evolution to Study Resistance Development
Purpose: To characterize the potential for and mechanisms of resistance development to new antibiotic candidates under controlled laboratory conditions [22].
Methodology (Adaptive Laboratory Evolution - ALE) [22]:
- Strain Selection: Select ancestral strains (both antibiotic-sensitive and MDR/XDR) of target pathogens.
- Experimental Evolution: Initiate multiple parallel-evolving populations (e.g., 10 per strain) in the presence of sub-inhibitory concentrations of the test antibiotic.
- Passaging and Concentration Increase: Passage populations regularly (e.g., daily) for an extended period (e.g., 60 days/120 generations), progressively increasing antibiotic concentration as resistance develops.
- Monitoring Resistance: Regularly assess MIC changes throughout the evolution experiment.
- Genetic Analysis: Sequence evolved lineages to identify mutations conferring resistance.
Diagram 1: Laboratory Evolution Workflow for Resistance Studies
Troubleshooting Guide: FAQs for Experimental Challenges
Q1: Our clinical isolates show inconsistent MIC results when tested repeatedly. What could be causing this variability?
A: Inconsistent MIC results can stem from several sources:
- Inoculum Density Variation: Ensure strict adherence to the 0.5 McFarland standard using calibrated spectrophotometry. Avoid using aged bacterial suspensions (>30 minutes after preparation).
- Antibiotic Stability: Verify proper storage conditions for antibiotic disks and powders. Some antibiotics like carbapenems are particularly labile.
- Agar Batch Variability: Use the same batch of Mueller-Hinton agar for comparable studies, as calcium and magnesium content can affect aminoglycoside and polymyxin activity.
- Heteroresistance: Consider that your isolate may contain subpopulations with differing resistance levels. Perform population analysis profiling to confirm.
Q2: We're unable to detect known carbapenemase genes in phenotypically resistant isolates. What alternative mechanisms should we investigate?
A: When genetic testing fails to explain phenotypic resistance, consider these alternative mechanisms:
- Porin Mutations: Loss or mutation of outer membrane porins (e.g., OprD in P. aeruginosa) combined with AmpC or ESBL production can confer carbapenem resistance without carbapenemases.
- Efflux Pump Overexpression: Upregulation of RND efflux systems (e.g., MexAB-OprM in P. aeruginosa, AdeABC in A. baumannii) can reduce intracellular antibiotic concentrations.
- Penicillin-Binding Protein (PBP) Modifications: Altered PBPs with reduced affinity for β-lactams.
- Biofilm Formation: Test for enhanced biofilm production which confers adaptive resistance.
Q3: Our novel compound shows excellent in vitro activity but fails in animal infection models. What could explain this discrepancy?
A: This common challenge in anti-infective development may result from:
- Protein Binding: High serum protein binding can significantly reduce free drug concentrations.
- Pharmacokinetics/Pharmacodynamics (PK/PD) Mismatch: The compound may not maintain concentrations above MIC for sufficient time at the infection site.
- Inoculum Effect: Efficacy may decrease at higher bacterial densities common in actual infections.
- Host Factor Interference: Host proteins or immune components may inactivate the compound.
- Biofilm Penetration: The compound may not effectively penetrate biofilms or host cells where bacteria reside.
Q4: We observe rapid resistance development to our novel antibiotic candidate in vitro. Should we abandon this compound?
A: Not necessarily. Rapid resistance development in laboratory evolution experiments doesn't always predict clinical failure but indicates the need for strategy adjustment [22]:
- Consider Combination Therapy: Test your compound with existing antibiotics for synergistic effects that suppress resistance emergence.
- Explore Dosing Optimization: Evaluate whether altered dosing regimens can suppress mutant selection.
- Assess Resistance Cost: Determine if resistance mutations incur fitness costs that limit their spread in absence of antibiotic pressure.
- Check for Pre-existing Resistance: Use functional metagenomics to assess whether resistance determinants already exist in environmental and human microbiomes [22].
Q5: How can we effectively test combination therapies against MDR Gram-negative pathogens?
A: For reliable combination testing:
- Use Checkerboard Assays: Prepare 2D serial dilutions of both antibiotics in microtiter plates to calculate Fractional Inhibitory Concentration (FIC) indices.
- Include Appropriate Controls: Always include single-agent controls and growth controls.
- Define Synergy Criteria: Use standardized definitions (e.g., FIC index ≤0.5 for synergy).
- Validate with Time-Kill Assays: Confirm static checkerboard results with dynamic time-kill studies that provide more clinically relevant data.
- Test Against Genetically Diverse Strains: Include multiple strains with different resistance mechanisms to ensure broad applicability.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagents for ESKAPE Pathogen Studies
| Reagent/Category | Specific Examples | Function/Application | Notes for Use |
|---|---|---|---|
| Culture Media | Mueller-Hinton Agar/Broth | Standardized AST | Must comply with CLSI performance specifications |
| Antibiotic Standards | CLSI-reference powders | MIC determination, QC | Verify purity and potency; proper storage critical |
| QC Strains | E. coli ATCC 25922, P. aeruginosa ATCC 27853 | Quality control for AST | Monitor for strain drift with repeated subculture |
| Molecular Detection Kits | PCR reagents for bla_KPC, bla_NDM, bla_OXA-48 | Rapid resistance gene detection | Include appropriate positive and negative controls |
| Efflux Pump Inhibitors | Phe-Arg-β-naphthylamide (PAβN) | Efflux mechanism studies | Cytotoxicity at high concentrations may limit use |
| Membrane Permeabilizers | Polymyxin B nonapeptide (PMBN) | Outer membrane studies | Lacks direct antibacterial activity |
| Biofilm Assessment Tools | Crystal violet, Calgary biofilm device | Biofilm quantification | Normalize to bacterial growth for accurate assessment |
Emerging Strategies to Overcome Intrinsic Resistance
The unique cell wall structure of Gram-negative bacteria represents both a key resistance mechanism and a potential "Achilles' heel" for therapeutic targeting [21]. Recent innovative approaches include:
Membrane Permeabilizers and Efflux Pump Inhibitors
Polymyxin Derivatives: Next-generation polymyxins like SPR741 and SPR206 show reduced nephrotoxicity while maintaining membrane-permeabilizing activity, making them ideal combination partners [21]. Octapeptins (e.g., octapeptin C4), structurally related to polymyxins, show promising activity against polymyxin-resistant strains with a lower propensity for resistance development [21].
Efflux Pump Inhibitors: While no clinical inhibitors are yet available, research continues to identify compounds that block major RND efflux systems, potentially resurrecting activity of existing antibiotics.
Alternative Therapeutic Modalities
Bacteriophage Therapy: Phages or phage-derived enzymes (endolysins) can specifically target resistant pathogens with minimal impact on commensal flora. Phage-antibiotic combinations demonstrate remarkable synergism both in vitro and in vivo [18].
Antimicrobial Peptides (AMPs): These naturally occurring host defense molecules often target bacterial membranes, making resistance development more difficult. Challenges remain in stabilizing AMPs in vivo and reducing production costs [16] [18].
Nanoparticles: Metal nanoparticles (e.g., silver, copper) can attack multiple cellular targets simultaneously, reducing the likelihood of resistance development. They can be used to create antimicrobial surface coatings for medical devices [16] [18].
Diagram 2: Strategies to Overcome Intrinsic Resistance
Antibiotic Adjuvants and Combination Approaches
Combining existing antibiotics with non-antibiotic adjuvants represents a promising strategy to extend the lifespan of current drugs. β-lactam/β-lactamase inhibitor combinations like ceftazidime-avibactam and meropenem-vaborbactam have successfully countered specific resistance mechanisms in clinical use [16]. Research continues to develop inhibitors targeting other resistance elements such as metallo-β-lactamases and efflux pumps.
The fight against WHO-critical ESKAPE pathogens requires continued innovation in both basic research methodologies and therapeutic development. By systematically addressing the unique challenges posed by intrinsic Gram-negative resistance and rapidly identifying emerging resistance mechanisms, the scientific community can develop more durable solutions to this pressing global health threat.
The rise of multidrug-resistant (MDR) Gram-negative bacteria represents one of the most severe threats to modern medicine, creating substantial clinical and economic burdens globally. Infections caused by these pathogens are associated with significantly higher treatment costs, extended hospital stays, and greater mortality rates compared to susceptible infections. The World Health Organization has classified several Gram-negative bacteria, including Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species, as critical priority pathogens due to their resistance profiles and impact on human health [15]. The economic ramifications extend far beyond individual patient care, with contemporary analyses revealing that antimicrobial resistance (AMR)-related healthcare costs exceed USD $100 billion annually globally, with projections indicating potential costs could rise to USD $300 billion by 2030 [23]. These figures encompass direct medical costs, increased length of hospital stays—averaging an additional 13 days for resistant infections—and the necessity for more expensive second-line antibiotics [23].
The fundamental structural differences in the cell envelopes of Gram-negative bacteria compared to Gram-positive species play a significant role in their intrinsic resistance to many antibiotic classes [15]. The Gram-negative cell envelope features an asymmetrical outer membrane with a lipopolysaccharide (LPS)-rich outer leaflet that acts as a formidable permeability barrier, effectively preventing access of hydrophobic molecules while porins limit diffusion of hydrophilic molecules to those below approximately 700 Da [15]. This combination confers intrinsic resistance to many antibiotic classes, including macrolides, glycopeptides, and lipopeptides, despite the fact that the targets of most antibiotics are highly conserved across Gram-positive and Gram-negative species [15]. Comprehending this economic and clinical burden is essential for directing research priorities and resource allocation toward innovative solutions for tackling MDR Gram-negative infections.
Quantitative Analysis of the Economic Burden
Global and Pathogen-Specific Cost Estimates
Recent comprehensive studies have quantified the staggering economic impact of antibiotic-resistant infections on healthcare systems worldwide. A 2025 analysis revealed that antibiotic resistance (ABR) was associated with a median value of $693 billion (IQR: $627 bn–$768 bn) in hospital costs globally, with productivity losses quantified at almost $194 billion annually [24]. The economic burden falls disproportionately on healthcare systems already struggling with limited resources, with low- and middle-income countries (LMICs) facing particular challenges due to less-effective antibiotics, limited access to healthcare, and poor infection practices [25].
Table 1: Hospital Costs Attributable to Antibiotic Resistance by Pathogen
| Pathogen/Resistance Type | Cost-per-Case Attributable to ABR | Notes |
|---|---|---|
| Multidrug-resistant Tuberculosis | $3,000 (lower-income) to $41,000 (high-income) | Highest mean hospital cost attributable to ABR per patient [24] |
| Carbapenem-resistant Infections | $3,000–$7,000 | Varies depending on syndrome [24] |
| General Bacterial AMR Infections | Up to $29,000 more per patient | Compared to susceptible infections [24] [26] |
The cost-per-case estimates reveal significant variations depending on the pathogen and resistance mechanism. Multidrug-resistant tuberculosis represents the most costly per patient, while carbapenem-resistant infections are also associated with substantial treatment expenses [24]. These elevated costs are driven by multiple factors including the need for more expensive antibiotics, longer hospitalization durations, and more intensive monitoring and supportive care requirements.
Regional Variations and Mortality Impact
The burden of MDR Gram-negative infections is not distributed evenly across global regions. Surveillance data from the WHO Global Antimicrobial Resistance Surveillance System (GLASS) reveals that resistance rates vary substantially by region, with particularly high prevalence in Southeast Asia and the Eastern Mediterranean [23]. Healthcare-associated infections caused by resistant organisms have increased by 35% since 2010, with particularly sharp rises observed in intensive care units and long-term care facilities [23]. The situation in developing nations presents additional challenges, where limited surveillance infrastructure, restricted access to newer antibiotics, and inadequate infection control measures contribute to higher resistance rates [23].
Table 2: Global Mortality and Regional Impact of AMR
| Region/Impact Measure | Statistics | Source/Timeframe |
|---|---|---|
| Global AMR-associated deaths (2019) | 4.95 million | [24] |
| Projected annual deaths by 2050 | 10 million | [26] |
| AMR mortality in Africa (2019) | 49% higher than HIV/AIDS and malaria combined | [25] |
| Healthcare-associated resistant infections | 67% increase in some regions | Since 2010 [23] |
| Community-acquired resistant infections | 38% increase in regions with high antibiotic misuse | [23] |
The mortality impact of AMR is particularly alarming in Africa, where it surpassed the combined mortality rate of HIV, AIDS, and malaria in 2019 [25]. Without effective intervention strategies, projections indicate AMR could lead to 8.22 million deaths associated with AMR and 1.91 million deaths directly attributable to it by 2050, with the highest all-age mortality rates expected to occur in South Asian, Latin American, and Caribbean countries [26].
Scientific Basis of Intrinsic Resistance in Gram-Negative Bacteria
Structural and Mechanistic Barriers
The formidable resilience of Gram-negative bacteria to multiple antibiotic classes stems primarily from their unique cell envelope architecture, which presents multiple barriers to antibiotic penetration and accumulation. The Gram-negative cell envelope consists of an inner cytoplasmic membrane, a thin peptidoglycan layer, and a distinctive asymmetric outer membrane containing lipopolysaccharide (LPS) in its outer leaflet [15]. This complex structure creates a sophisticated permeability barrier that limits antibiotic access to intracellular targets.
The LPS layer effectively prevents access of hydrophobic molecules, while porins—transmembrane β-barrel proteins—mediate the uptake of small hydrophilic molecules but restrict passage to those below approximately 700 Da [15]. Beyond this passive barrier, Gram-negative bacteria employ active efflux systems that recognize and export a broad spectrum of antibiotics back across the outer membrane, further reducing intracellular drug accumulation [15]. These combined mechanisms confer intrinsic resistance to many antibiotic classes including macrolides, glycopeptides, and lipopeptides, despite the conservation of their cellular targets across bacterial species [15].
Key Resistance Mechanisms in Priority Pathogens
The clinical significance of specific resistance mechanisms varies among priority Gram-negative pathogens. Carbapenem-resistant Enterobacteriaceae (CRE), particularly Klebsiella pneumoniae, often employ carbapenemase enzymes that hydrolyze these last-resort β-lactam antibiotics [23]. Acinetobacter baumannii exhibits remarkable genetic plasticity, acquiring resistance genes through horizontal gene transfer and upregulating efflux systems [3]. Pseudomonas aeruginosa utilizes its inherently low outer membrane permeability combined with inducible resistance mechanisms to resist multiple antibiotic classes [3] [15].
Polymyxin resistance represents a particularly concerning development, mediated through multiple mechanisms including the modification of lipid A components of LPS via chromosomal mutations or mobile colistin resistance (mcr) genes [3]. The mcr-1 gene, originally described in Escherichia coli and now disseminated globally on highly transmissible plasmids, encodes a phosphoethanolamine transferase that modifies lipid A, reducing the negative charge of LPS and decreasing polymyxin binding [3]. In A. baumannii, complete loss of LPS through mutations in lpxACD genes represents another pathway to polymyxin resistance [3].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Gram-Negative Resistance
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Membrane Permeabilizers | Polymyxin B nonapeptide (PMBN), SPR741, SPR206, Octapeptin C4 | Outer membrane disruption to enhance antibiotic penetration [3] [15] |
| Efflux Pump Inhibitors | Phe-Arg-β-naphthylamide (PAβN), MC-207,110 | Block multidrug efflux systems to increase intracellular antibiotic accumulation [15] |
| LPS Biosynthesis Inhibitors | Dephostatin, LpxC inhibitors | Target lipopolysaccharide synthesis to compromise outer membrane integrity [3] |
| β-Lactamase Inhibitors | Avibactam, Vaborbactam, Relebactam | Counteract enzymatic degradation of β-lactam antibiotics [26] [15] |
| Two-Component System Inhibitors | PmrAB and PhoPQ pathway inhibitors | Disrupt regulatory networks controlling resistance gene expression [3] |
| Chemical Libraries | CC4CARB compound collections | Source of novel scaffolds for antibacterial development [27] |
The CC4CARB (Chemistry Center for Combating Antibiotic Resistant Bacteria) initiative represents an important resource for researchers, providing access to specialized chemical libraries designed to overcome the unique challenges of Gram-negative antibiotic discovery [27]. These collections contain compounds with structural features favorable for penetration through Gram-negative outer membranes, addressing a critical gap in conventional screening libraries that are often tailored to mammalian cell targets [27].
Troubleshooting Guides & FAQs: Addressing Experimental Challenges
Troubleshooting Membrane Permeabilization Studies
Problem: Inconsistent results in outer membrane permeabilization assays using adjuvant compounds.
- Potential Cause 1: Variable compound stability or solubility.
- Solution: Prepare fresh compound solutions for each experiment using appropriate solvents. Verify solubility visually and quantify concentration using validated analytical methods (e.g., HPLC). Include stability tests under experimental conditions.
- Potential Cause 2: Strain-to-strain differences in outer membrane composition.
- Potential Cause 3: Inadequate controls leading to misinterpretation.
- Solution: Implement a complete set of controls including (1) bacteria without antibiotic, (2) bacteria with antibiotic alone, (3) bacteria with adjuvant alone, and (4) a known permeabilizer (e.g., PMBN) as a positive control [28].
Problem: Poor synergy between membrane permeabilizers and partner antibiotics in checkerboard assays.
- Potential Cause 1: Incompatible mechanisms of action.
- Solution: Research the entry requirements of the partner antibiotic. Verify that permeabilization adequately addresses the primary barrier for that specific antibiotic (e.g., porin pathway vs. self-promoted uptake) [15].
- Potential Cause 2: Efflux pump activity expelling the antibiotic.
- Solution: Incorporate an efflux pump inhibitor in parallel experiments. Use strains with deleted efflux pump components to dissect the contribution of efflux versus permeability [15].
- Potential Cause 3: Antibiotic degradation by bacterial enzymes.
- Solution: Test antibiotic stability in culture medium alone and in the presence of bacteria. Consider adding enzyme inhibitors specific to known resistance mechanisms (e.g., β-lactamase inhibitors) [15].
FAQs on Gram-Negative Resistance Research
Q: What are the "eNTRy Rules" and how can they guide my compound design? A: The "eNTRy Rules" are a set of guiding principles developed by Richter and Hergenrother for compound accumulation in Escherichia coli [15]. They provide predictive parameters for designing compounds capable of penetrating Gram-negative bacteria, focusing on properties like molecular weight, polarity, and charge. These rules are particularly valuable for optimizing Gram-positive-active compounds to expand their spectrum to include Gram-negative pathogens.
Q: Why is heteroresistance a significant problem in polymyxin treatment, and how can I detect it in my experiments? A: Heteroresistance occurs when a susceptible bacterial population contains a resistant subpopulation that can emerge during treatment, leading to therapeutic failure [3]. This phenomenon is particularly problematic for polymyxins and is often associated with mutations in the pmrCAB operon or other regulatory genes [3]. To detect heteroresistance, perform population analysis profiling (PAP), where a large bacterial inoculum (≥10^9 CFU) is plated on antibiotic-containing plates. The growth of resistant colonies at antibiotic concentrations above the MIC indicates heteroresistance.
Q: What are the most promising regulatory targets for overcoming intrinsic resistance? A: Two-component systems (TCS) that regulate membrane stress responses represent promising targets. Specifically:
- PmrAB/PhoPQ systems: Regulate LPS modification genes (pmrC, arn operon) involved in polymyxin resistance [3].
- Regulators of efflux pump expression: Control multidrug efflux systems that contribute to intrinsic resistance [15]. Small molecule inhibitors targeting these regulatory networks can potentially resensitize bacteria to existing antibiotics.
Q: How can I effectively test novel compounds for activity against Gram-negative bacteria with intact permeability barriers? A: Utilize a combination of approaches:
- Use hypersensitive strains: Include strains with permeabilized outer membranes (e.g., E. coli DC2) or deleted efflux pumps to distinguish between intrinsic activity and permeability issues [15].
- Employ permeabilizing adjuvants: Test compounds in combination with sub-inhibitory concentrations of membrane permeabilizers like PMBN to assess intrinsic antibacterial activity once the permeability barrier is compromised [15].
- Check accumulation directly: Use LC-MS/MS methods to directly measure intracellular compound accumulation in wild-type versus membrane-compromised strains [15].
The economic and clinical burden of multidrug-resistant Gram-negative infections continues to escalate, demanding innovative approaches to antibiotic discovery and development. The current antibacterial pipeline remains insufficient to address the increasing prevalence of resistant infections, with only 12 of 32 traditional agents targeting WHO priority pathogens meeting innovation criteria [26]. Encouragingly, vaccines against key pathogens like Staphylococcus aureus, Escherichia coli, and Klebsiella pneumoniae could potentially avert 30-40% of hospital costs and productivity losses associated with antibiotic resistance according to recent modeling studies [24].
Future success in combating MDR Gram-negative infections will require a multi-pronged strategy including:
- Adjuvant development to potentiate existing antibiotics by overcoming specific resistance mechanisms [3] [15]
- Novel compound discovery focusing on entirely new targets and chemical scaffolds [27]
- Vaccine development to prevent infections caused by high-burden resistant pathogens [24]
- Enhanced diagnostics to enable rapid detection of resistance mechanisms and appropriate antibiotic selection [23]
- Global collaboration through initiatives like CC4CARB that address the economic challenges of antibiotic development [27]
The significant economic burden quantified in recent studies underscores the urgent need for increased investment and coordinated global action to address the threat of multidrug-resistant Gram-negative bacteria. By integrating mechanistic understanding of resistance with innovative therapeutic approaches and robust economic analysis, the scientific community can develop effective strategies to overcome these formidable pathogens.
Breaching the Defenses: Cutting-Edge Therapeutic Strategies and Applications
The intrinsic resistance of Gram-negative bacteria constitutes a formidable barrier in antimicrobial therapy, primarily due to their unique cell envelope structure comprising a dual-membrane system. This architecture significantly reduces membrane permeability and facilitates active drug efflux, rendering many conventional antibiotics ineffective [29] [30]. The escalating crisis of antimicrobial resistance (AMR), directly responsible for 1.27 million global deaths annually with a contribution to 4.95 million deaths, underscores the urgent need for innovative therapeutic strategies [31]. Among the most critical pathogens identified by the World Health Organization are Gram-negative bacteria such as Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter baumannii, which exhibit alarming resistance rates to last-resort antibiotics including carbapenems [32] [31].
Antibiotic adjuvants represent a promising approach to circumvent existing resistance mechanisms without directly killing bacteria themselves. These compounds, when co-administered with conventional antibiotics, can potentiate their activity by targeting bacterial defense systems [33] [34]. The three primary classes of adjuvants—β-lactamase inhibitors, efflux pump inhibitors, and outer membrane permeabilizers—function through distinct mechanisms to restore antibiotic efficacy against resistant strains. This technical support center provides detailed guidance for researchers developing these critical compounds, with a specific focus on overcoming intrinsic resistance in Gram-negative pathogens.
Troubleshooting Guides and FAQs
β-Lactamase Inhibitors
FAQ: Why does my β-lactam/β-lactamase inhibitor combination show poor efficacy against clinical isolates despite in vitro susceptibility?
Answer: This discrepancy often arises from several technical and biological factors:
- Inoculum Effect: High bacterial densities can overwhelm inhibitor capacity. Repeat testing with standardized inoculum (1-5 × 10^5 CFU/mL) [34].
- Extended-Spectrum β-Lactamase (ESBL) Variants: Your inhibitor may not cover certain ESBL classes (e.g., CTX-M, PER, VEB). Characterize the specific β-lactamase genes present in your isolates using PCR or whole-genome sequencing [29].
- Porin Deficiencies: Reduced outer membrane permeability limits intracellular antibiotic accumulation. Check for porin mutations (e.g., OmpF/OmpC in E. coli, OprD in P. aeruginosa) [29] [30].
- Efflux Pump Overexpression: Concurrent efflux activity can expel both antibiotic and inhibitor. Consider adding an efflux pump inhibitor to your assay [30].
Troubleshooting Guide: Inhibitor Restoration of β-Lactam Activity
| Observation | Potential Cause | Suggested Solution |
|---|---|---|
| No restoration of antibiotic activity | Irreversible inhibitor binding | Test serine β-lactamase inhibitors (e.g., avibactam, relebactam, vaborbactam) [33] |
| Partial restoration of activity | Metallo-β-lactamases (MBLs) present | MBLs are not inhibited by conventional inhibitors; consider alternative strategies [29] |
| Variable activity across strains | Multiple β-lactamase classes | Use inhibitor combinations or broad-spectrum inhibitors [30] |
| Initial efficacy followed by resistance | Selection of resistant mutants | Check for inhibitor resistance mutations (e.g., K234R substitution in KPC) |
Experimental Protocol: Time-Kill Assay for β-Lactam/Inhibitor Combinations
- Preparation: Grow bacterial overnight culture in cation-adjusted Mueller-Hinton broth (CAMHB) to mid-log phase (OD600 ≈ 0.5).
- Standardization: Adjust suspension to 1 × 10^6 CFU/mL in fresh CAMHB.
- Treatment: Add antibiotic alone, inhibitor alone, and combination at clinically relevant concentrations. Include growth control.
- Incubation: Incubate at 35±2°C with shaking. Remove aliquots at 0, 4, 8, and 24 hours.
- Quantification: Serially dilute aliquots, plate on appropriate agar, and enumerate colonies after 18-24 hours incubation.
- Analysis: Plot log10 CFU/mL versus time. Synergy is defined as ≥2-log10 decrease with combination compared to most active single agent [33] [30].
Efflux Pump Inhibitors
FAQ: How can I distinguish between efflux-mediated resistance and other resistance mechanisms in Gram-negative bacteria?
Answer: Implement a systematic approach:
- Ethidium Bromide Accumulation Assay: Compare intracellular ethidium bromide fluorescence with/without efflux pump inhibitor (e.g., PaβN, CCCP). Increased fluorescence with inhibitor suggests active efflux [30].
- Checkerboard Synergy Testing: Test antibiotic with known efflux pump inhibitors. Fractional Inhibitory Concentration (FIC) index ≤0.5 indicates synergy and potential efflux involvement [30] [34].
- Gene Expression Analysis: Quantify expression of efflux pump genes (e.g., acrAB, mexAB, adeABC) via RT-qPCR. Overexpression (>4-fold increase) suggests efflux contribution [29].
- Control Strains: Include efflux-deficient mutants (e.g., ΔacrB) as controls to confirm efflux-mediated resistance [30].
Troubleshooting Guide: Efflux Pump Inhibition Challenges
| Observation | Potential Cause | Suggested Solution |
|---|---|---|
| Cytotoxicity of inhibitor | Non-selective targeting of mammalian cells | Optimize chemical structure for selective bacterial target engagement [34] |
| Poor potentiation in vivo | Pharmacokinetic mismatch with antibiotic | Align dosing schedules or develop co-formulation [33] |
| Species-specific activity | Differential efflux pump expression/structure | Validate across multiple target pathogens [29] |
| Rapid resistance development | Single-target mechanism | Develop multi-target inhibitors or combination adjuvant approaches [30] |
Experimental Protocol: Ethidium Bromide Accumulation Assay
- Cell Preparation: Grow bacteria to mid-log phase, harvest by centrifugation (3,500 × g, 10 min), and wash twice with PBS.
- Loading: Resuspend cells to OD600 = 0.2 in PBS containing 20 µM ethidium bromide.
- Treatment: Divide suspension into aliquots with/without efflux pump inhibitor (e.g., 50 µM PaβN).
- Measurement: Transfer to black-walled microplates and monitor fluorescence (excitation 530 nm, emission 600 nm) every 2-5 minutes for 60 minutes at 37°C.
- Analysis: Calculate accumulation rate from linear portion of fluorescence versus time curve. Compare initial rates with/without inhibitor [30].
Outer Membrane Permeabilizers
FAQ: Why do some permeabilizers show excellent in vitro activity but fail in animal models?
Answer: This translational gap often results from:
- Serum Binding: High protein binding reduces free drug concentration. Determine protein binding percentage and adjust dosing [33].
- Toxicity Limitations: Membrane disruption can affect eukaryotic cells at higher doses. Conduct thorough cytotoxicity screening (e.g., hemolysis, hepatotoxicity) early in development [30].
- Pharmacokinetic/Pharmacodynamic (PK/PD) Mismatch: Differing half-lives between permeabilizer and antibiotic. Design permeabilizers with compatible PK profiles [33].
- In Vivo Compensation: Bacterial adaptation through increased efflux or alternative membrane modifications. Test resistance development potential through serial passage experiments [30].
Troubleshooting Guide: Membrane Permeabilization Issues
| Observation | Potential Cause | Suggested Solution |
|---|---|---|
| Increased antibiotic uptake but no efficacy | Intracellular enzymatic degradation | Combine with enzyme inhibitors [30] |
| Species-specific permeabilization | Differential LPS structure | Tailor permeabilizers to target pathogen LPS composition [29] |
| Synergy with large antibiotics only | Size-selective porin formation | Optimize molecular size/shape for broader antibiotic coverage [30] |
| Disruption of mammalian membranes | Lack of selectivity for bacterial membranes | Modify chemical structure to target LPS-specific interactions [34] |
Experimental Protocol: Outer Membrane Permeability Assessment Using NPN Assay
- Reagent Preparation: Prepare 10 µM N-phenyl-1-naphthylamine (NPN) in 5 mM HEPES buffer (pH 7.2).
- Cell Preparation: Harvest mid-log phase bacteria, wash twice with 5 mM HEPES (pH 7.2), and adjust to OD600 = 0.5 in the same buffer.
- Measurement: Add 10 µL of NPN solution to 190 µL of cell suspension in black-walled microplates.
- Treatment: Add permeabilizer at sub-MIC concentrations and immediately measure fluorescence (excitation 350 nm, emission 420 nm) every 30 seconds for 10 minutes.
- Analysis: Calculate maximum fluorescence increase and initial rate of fluorescence change. Compare with positive control (e.g., polymyxin B) and untreated cells [30].
Data Presentation
Quantitative Comparison of Major Adjuvant Classes
Table 1: Comparative Analysis of Antibiotic Adjuvant Classes
| Parameter | β-Lactamase Inhibitors | Efflux Pump Inhibitors | Outer Membrane Permeabilizers |
|---|---|---|---|
| Molecular Targets | Serine β-lactamases (e.g., TEM, SHV, CTX-M, KPC); Some MBLs (e.g., VIM, NDM) [29] | RND-type pumps (e.g., AcrAB-TolC, MexAB-OprM, AdeABC) [30] | Lipopolysaccharide (LPS) layer; Cationic bridges between LPS molecules [30] |
| Resistance Mechanisms | Mutations in active site; Alternative hydrolases; Overexpression [29] | Target site mutations; Overexpression of alternative pumps; Regulatory mutations [30] | LPS modification (e.g., lipid A phosphorylation); Cationic substitution; Efflux upregulation [29] |
| Clinical Status | Multiple approved (clavulanate, tazobactam, avibactam, vaborbactam) [33] | Limited clinical approval (none widely used); Mostly preclinical development [30] | Polymyxin derivatives approved; Novel agents in development [30] |
| Potentiation Spectrum | Primarily β-lactams (penicillins, cephalosporins, carbapenems) [33] | Broad-spectrum (multiple antibiotic classes) [30] | Broad-spectrum, especially against Gram-negative pathogens [30] |
| Key Challenges | MBL inhibition; Inhibitor-resistant variants [29] | Host toxicity; Pharmacokinetic optimization [30] | Specificity for bacterial membranes; Toxicity concerns [34] |
Research Reagent Solutions
Table 2: Essential Research Reagents for Adjuvant Development
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Reference Strains | E. coli ATCC 25922; P. aeruginosa PAO1; K. pneumoniae ATCC 13883; A. baumannii ATCC 19606 [29] | Method standardization; Quality control | Include clinical MDR isolates for translational relevance [30] |
| Control Inhibitors | Clavulanic acid (β-lactamase); Phenylalanine-arginine β-naphthylamide (PaβN, efflux); Polymyxin B nonapeptide (permeabilizer) [30] [34] | Assay validation; Comparator studies | Use clinically relevant concentrations based on pharmacokinetic data [33] |
| Specialized Media | Cation-adjusted Mueller-Hinton broth; RPMI-1640 for biofilm studies [30] | Standardized susceptibility testing; Biofilm models | Adjust calcium/magnesium concentrations for polymyxin testing [30] |
| Molecular Tools | β-lactamase nitrocefin assay; Ethidium bromide accumulation; N-phenyl-1-naphthylamine (NPN) uptake [30] | Mechanism confirmation; High-throughput screening | Validate with appropriate controls and standard curves [30] |
| Animal Models | Mouse thigh infection; Neutropenic lung infection; Sepsis models [33] | In vivo efficacy assessment | Consider immune status and inoculation method for clinical relevance [33] |
Visualization of Mechanisms and Workflows
Adjuvant Mechanisms Against Gram-Negative Resistance
Diagram 1: Adjuvant action mechanisms on Gram-negative bacterial cell
High-Throughput Adjuvant Screening Workflow
Diagram 2: Adjuvant screening and development workflow
The strategic deployment of antibiotic adjuvants represents a critical approach to extending the therapeutic lifespan of existing antibiotics against multidrug-resistant Gram-negative pathogens. As research advances, the integration of structural biology, computational design, and sophisticated screening methodologies will accelerate the development of next-generation adjuvants with enhanced potency and reduced susceptibility to resistance. The experimental frameworks and troubleshooting guides provided herein offer practical resources for researchers navigating the technical challenges inherent in this vital field of antimicrobial discovery. Through continued innovation in adjuvant technology, the scientific community can mount a more effective defense against the escalating threat of Gram-negative resistance.
This technical support guide addresses the application of membrane-targeting Antimicrobial Peptides (AMPs) and their mimetics in research focused on overcoming intrinsic resistance in Gram-negative bacteria. The unique structure of the Gram-negative outer membrane (OM), featuring an asymmetrical lipopolysaccharide (LPS)-rich bilayer, serves as a formidable permeability barrier, conferring intrinsic resistance to many conventional antibiotics [10] [35] [36]. Membrane-targeting agents represent a promising therapeutic strategy as they can disrupt the integrity of this essential cellular structure, leading to increased membrane permeability, depolarization, and ultimately, bacterial cell death [37] [38]. This document provides troubleshooting guides, FAQs, and experimental protocols to support researchers in this critical field.
Frequently Asked Questions (FAQs) on Mechanisms and Resistance
Q1: Why are Gram-negative bacteria intrinsically more resistant to many antibiotics than Gram-positive bacteria? The primary reason is the presence of a complex, asymmetrical outer membrane (OM) in Gram-negative bacteria. This OM has an inner leaflet of phospholipids and an outer leaflet composed predominantly of lipopolysaccharide (LPS) [10] [35] [36]. The dense, polyanionic nature of LPS, stabilized by divalent cations (Mg²⁺, Ca²⁺), creates a highly impermeable barrier to hydrophobic molecules and large antibiotics [10] [35]. Furthermore, the passage of hydrophilic molecules is restricted to porin channels, which are size-selective (typically <700 Da) [10] [3]. This combination of a formidable LPS barrier and selective porins significantly limits the intracellular accumulation of many antibiotics.
Q2: What is the primary mechanism of action for most Antimicrobial Peptides (AMPs) against Gram-negative bacteria? Most AMPs are cationic and exert their activity through initial electrostatic interactions with the negatively charged components of the bacterial membrane, such as the phosphate groups on LPS [37] [39]. This is followed by integration into the membrane bilayer, leading to physical disruption. The specific models of disruption include:
- The "Carpet" Model: Peptides cover the membrane surface like a carpet, leading to membrane thinning and eventual micellization or the formation of large holes.
- Toroidal Pore Model: Peptides insert into the membrane, inducing lipid monolayers to curve continuously, forming a pore lined by both peptide and lipid headgroups.
- Barrel-Stave Model: Peptides assemble into a barrel-like structure within the membrane, with the hydrophobic regions facing the lipids and the hydrophilic regions forming an internal channel [37] [38]. This physical disruption compromises membrane integrity, causing depolarization, leakage of cellular contents, and cell death.
Q3: What are the common resistance mechanisms Gram-negative bacteria employ against membrane-targeting agents? Bacteria have evolved several mechanisms to resist AMPs and mimetics:
- Modification of Membrane Components: The most common mechanism involves the enzymatic modification of lipid A, the core component of LPS, to reduce its net negative charge. This is achieved by the addition of phosphoethanolamine or 4-amino-4-deoxy-L-arabinose (L-Ara4N) via systems like PmrAB and arnBCADTEF, which decreases electrostatic interaction with cationic AMPs [36] [3].
- Efflux Pumps: Resistance-Nodulation-Division (RND) family efflux pumps can actively export a wide range of compounds, including some AMPs, from the periplasm and cytoplasm back to the extracellular environment [35] [40].
- Membrane Vesicle Shedding: Bacteria can release outer membrane vesicles (OMVs) that encapsulate and sequester antimicrobial agents, effectively removing them from the cell surface [35].
- Proteolytic Degradation: Production of extracellular proteases can lead to the degradation of peptide-based agents before they reach their target [37].
Q4: What are antibiotic adjuvants and how can they help overcome resistance? Antibiotic adjuvants are non-microbicidal compounds that enhance the efficacy of co-administered antibiotics. In the context of membrane-targeting, adjuvants like polymyxin B nonapeptide (PMBN) and its analogs (e.g., SPR741) permeabilize the outer membrane but lack significant direct antibacterial activity. By disrupting the OM, they facilitate the entry of other antibiotics into the cell, re-sensitizing resistant bacteria to drugs to which they were previously impermeable [10] [3].
Troubleshooting Common Experimental Issues
Problem: High Cytotoxicity and Hemolytic Activity of AMPs in Mammalian Cell Assays.
- Potential Cause: The AMP has low selectivity for bacterial over mammalian membranes, often due to a high hydrophobic moment or non-optimal charge.
- Solutions:
- Sequence Optimization: Utilize rational design or deep learning models (e.g., deepAMP) to optimize sequences for higher specificity. Incorporating D-amino acids can increase stability against proteases and potentially reduce cytotoxicity [37] [41].
- Check Physicochemical Parameters: Aim for a net charge between +2 and +9 and a hydrophobicity that balances activity and selectivity. Tools like the APD3 database can provide guidance.
- Utilize Nanocarriers: Employ lipid-based or polymeric nanoparticles to deliver AMPs. This can enhance target specificity, reduce systemic exposure, and lower cytotoxic effects [42].
Problem: Low Antimicrobial Activity of a Novel AMP Against Clinical Isolates.
- Potential Causes:
- Pre-existing bacterial resistance mechanisms (see FAQ Q3).
- Inoculum size is too high.
- Peptide degradation during storage or assay.
- Cationic antagonism from the growth medium.
- Solutions:
- Use Standardized Inoculum: Ensure a consistent and recommended inoculum size (e.g., ~5x10⁵ CFU/mL).
- Change Media: Use low-salt buffers or media like Mueller-Hinton broth, which is standardized for antimicrobial susceptibility testing. High salt concentrations can inhibit electrostatic interactions.
- Include Protease Inhibitors: If peptide degradation is suspected, add protease inhibitors to the storage buffer, or use peptides with stabilized structures (e.g., cyclized).
- Test Against Defined Mutants: Use laboratory strains with known mutations (e.g., in pmrAB or arn genes) to identify specific resistance mechanisms at play [3].
Problem: Inconsistent Results in Membrane Depolarization Assays.
- Potential Causes: Inconsistent dye loading, improper calibration, or interference from the test compound.
- Solutions:
- Control Experiments: Always include a vehicle control and a control with a known depolarizing agent (e.g., gramicidin).
- Dye Concentration Titration: Titrate the concentration of the fluorescent dye (e.g., DiSC₃(5)) to ensure optimal loading without self-quenching.
- Wavelength Verification: Confirm that the test compound does not auto-fluoresce or quench the dye at the monitored wavelengths.
Quantitative Data on Selected AMPs and Mimetics
The following table summarizes key quantitative data for selected membrane-targeting agents, including those in clinical development. MIC (Minimum Inhibitory Concentration) and MHC (Minimum Hemolytic Concentration) are critical for assessing potency and selectivity.
Table 1: Activity and Selectivity Profiles of Key Membrane-Targeting Agents
| Agent Name | Status / Class | Gram-negative MIC (μM) | Gram-positive MIC (μM) | Haemolysis | Therapeutic Index (MHC/MIC) Estimate | Primary Mechanism |
|---|---|---|---|---|---|---|
| Colistin (Polymyxin E) [37] | FDA-approved, Natural AMP | ≤1.7 μM | - | 0–1.8% at 0.12 μg/mL | High (clinical use) | Membranolytic, LPS binding |
| Pexiganan (MSI-78) [37] | Phase III, Designed AMP | 3.23–6.46 μM | 3.23–12.9 μM | 5–63% at 50–64 μg/mL | Low | Toroidal pore formation |
| LL-37 [37] | Phase II, Human Cathelicidin | 0.04–16 μM | 0.16–16 μM | 1.5–5% in MIC range | Low | Membranolytic, immunomodulatory |
| SPR741 [3] | Phase I, Adjuvant | Lacks direct activity | Lacks direct activity | Low (in models) | N/A (Adjuvant) | Outer Membrane Permeabilization |
| T2-9 (deepAMP) [41] | Pre-clinical, AI-designed | Comparable to FDA-approved antibiotics (specific values not provided) | Not specified | Low (inferred from study) | High (inferred) | Membrane disruption |
Table 2: Common Research Reagents for Studying Membrane-Targeting Agents
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| Polymyxin B Nonapeptide (PMBN) [10] [3] | Outer membrane permeabilizer; used as an adjuvant to potentiate other antibiotics in research. | Benchmark compound for studying OM disruption without direct killing. |
| DiSC₃(5) [38] | Fluorescent dye for measuring membrane potential (depolarization). | Signal increases upon membrane depolarization and dye release into medium. |
| N-Phenyl-1-naphthylamine (NPN) [10] | Hydrophobic fluorescent probe for assessing outer membrane permeability. | Fluorescence increases in a compromised OM. |
| Lauryl Tryptose Broth | Low-salt growth medium for AMP susceptibility testing. | Reduces cationic antagonism, providing a more accurate assessment of AMP activity. |
| Lipopolysaccharide (LPS) | Used in surface plasmon resonance (SPR) or other biophysical assays to study initial binding kinetics of AMPs. | Isolated from various Gram-negative species (e.g., E. coli, P. aeruginosa). |
| Large Unilamellar Vesicles (LUVs) | Synthetic membrane models for studying lipid-peptide interactions and mechanisms of membrane disruption. | Can be prepared with defined lipid compositions (e.g., POPG:POPE to mimic bacterial membranes). |
Detailed Experimental Protocols
Protocol 1: Minimum Inhibitory Concentration (MIC) Assay (Broth Microdilution)
Principle: This standard broth microdilution method determines the lowest concentration of an antimicrobial agent that inhibits visible growth of a microorganism. Materials:
- Cation-adjusted Mueller-Hinton Broth (CAMHB)
- Sterile 96-well polypropylene microtiter plates
- Bacterial inoculum (adjusted to ~5x10⁵ CFU/mL)
- Test compound (serial dilutions prepared in CAMHB) Method:
- Prepare two-fold serial dilutions of the AMP/mimetic in CAMHB across the microtiter plate (e.g., 100 μL/well).
- Inoculate each well with 100 μL of the standardized bacterial suspension. Include growth control (bacteria + CAMHB) and sterility control (CAMHB only) wells.
- Seal the plate and incubate at 37°C for 16-20 hours without shaking.
- The MIC is the lowest concentration of the agent that completely inhibits visible growth.
Protocol 2: Outer Membrane Permeabilization Assay Using NPN
Principle: The fluorescent probe NPN is quenched in aqueous environments but exhibits enhanced fluorescence in a hydrophobic environment. A disrupted outer membrane allows NPN to enter and intercalate into the phospholipid inner membrane, causing a measurable increase in fluorescence [10]. Materials:
- Bacterial cells in mid-logarithmic phase
- 5 mM HEPES buffer (pH 7.2) containing 5 mM glucose
- NPN stock solution (0.5 mM in acetone)
- Test compound
- Fluorescence plate reader or spectrophotometer Method:
- Harvest and wash bacterial cells, then resuspend in HEPES-glucose buffer to an OD₆₀₀ of ~0.5.
- Mix 200 μL of cell suspension with 10 μL of NPN (final concentration 10 μM) in a well of a black microtiter plate.
- Add the test compound and immediately monitor fluorescence (excitation: 350 nm, emission: 420 nm) over time (e.g., 30 minutes).
- Include a positive control (e.g., PMBN) and a negative control (buffer only). The rate and extent of fluorescence increase indicate the degree of OM permeabilization.
Protocol 3: Membrane Depolarization Assay Using DiSC₃(5)
Principle: The DiSC₃(5) dye accumulates in the polarized cytoplasmic membrane, where it is self-quenched. Membrane disruption or pore formation by an antimicrobial agent causes depolarization, leading to the release of the dye and a consequent increase in fluorescence [38]. Materials:
- Bacterial cells in mid-logarithmic phase
- Assay buffer (e.g., 5 mM HEPES, 20 mM glucose, pH 7.4)
- DiSC₃(5) stock solution (1 mM in DMSO)
- Test compound
- Fluorescence plate reader Method:
- Wash and resuspend bacterial cells in assay buffer to an OD₆₀₀ of ~0.05.
- Load the cells with DiSC₃(5) (final concentration 0.4 μM) for ~30-60 minutes until the fluorescence signal stabilizes (quenched state).
- Dispense the dye-loaded cells into a microtiter plate. Add the test compound and monitor fluorescence immediately (excitation: 622 nm, emission: 670 nm).
- A positive control (e.g., gramicidin) that fully depolarizes the membrane should be included. The increase in fluorescence is proportional to the degree of membrane depolarization.
Visual Workflows and Diagrams
Diagram 1: Mechanism of AMP Action and Bacterial Resistance
Diagram 2: Experimental Workflow for AMP Characterization
Combating multidrug-resistant Gram-negative bacterial infections represents one of the most critical global health threats today. These pathogens, including Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae such as Escherichia coli and Klebsiella pneumoniae, possess a complex cell envelope that significantly complicates treatment with conventional antibiotics [8]. Their intrinsic resistance stems from several sophisticated mechanisms:
- Impermeable Outer Membrane: The asymmetric lipid bilayer structure, featuring lipopolysaccharides (LPS) in the outer leaflet, creates a formidable permeability barrier [8].
- Efflux Pump Systems: Membrane proteins actively extrude antibiotics before they reach intracellular targets, with systems like AcrB in E. coli and MexB in P. aeruginosa being particularly effective [43].
- Enzymatic Degradation: Production of β-lactamases (including ESBL, AmpC, and carbapenemases) hydrolyzes and inactivates antibiotic compounds [44].
- Biofilm Formation: Matrix-enclosed bacterial communities present a physical barrier that delays antibiotic penetration and fosters persistent infections [43].
Antimicrobial peptides (AMPs) offer a promising therapeutic alternative due to their rapid bactericidal effects, multiple mechanisms of action, and lower potential for inducing resistance compared to conventional antibiotics [42]. However, their clinical application is hampered by limitations including enzymatic degradation, cytotoxicity, poor pharmacokinetic properties, and high production costs [42] [45].
Nanocarrier-based delivery systems present an innovative strategy to overcome these challenges by enhancing peptide stability, bioavailability, and targeted delivery while reducing systemic toxicity [42]. This technical support center provides researchers with practical guidance for implementing these advanced systems in their work against Gram-negative pathogens.
FAQs: Addressing Key Technical Challenges
Q1: What are the primary advantages of using nanocarriers for AMP delivery against Gram-negative bacteria?
Nanocarriers enhance AMP efficacy through multiple mechanisms:
- Enhanced Stability: Polymeric matrices protect AMPs from proteolytic degradation in the harsh physiological environment, significantly extending their half-life [43]. For example, chitosan nanoparticles successfully protected cryptdin-2 against gastrointestinal degradation, increasing survival rates from 0% to 83% in a mouse model of Salmonella infection [43].
- Improved Bioavailability: Nano-encapsulation increases water solubility of poorly soluble antimicrobial compounds. Supramolecular hydrogel nano-antibiotics increased triclosan solubility by 850-fold while enabling targeted accumulation at infection sites [43].
- Overcoming Resistance Mechanisms: Nanocarriers can bypass efflux pumps and outer membrane barriers through endocytic pathways, delivering higher intracellular antibiotic concentrations [43]. Coating penicillin G in polyacrylate nanoparticles rejuvenated its activity against β-lactamase-producing MRSA by protecting it from enzymatic degradation [43].
- Reduced Toxicity: Encapsulation minimizes undesirable interactions between AMP cationic residues and host cells. Colistin-encapsulated micelles exhibited a maximum tolerated dose in mice at least 16 times higher than the free drug while preserving bactericidal activity [43].
Q2: Which nanocarrier systems show the most promise for AMP delivery?
Research indicates several effective nanocarrier platforms, each with distinct advantages:
Table 1: Promising Nanocarrier Systems for AMP Delivery
| Nanocarrier Type | Key Advantages | Demonstrated Efficacy | Considerations |
|---|---|---|---|
| Polymeric Nanoparticles (PLGA, PCL, Chitosan) | Biocompatible, biodegradable, controlled release profiles | Chitosan NPs for cryptdin-2 delivery improved survival in Salmonella-infected mice [43] | Batch-to-batch variability during synthesis |
| Lipid-Based Nanoparticles | High encapsulation efficiency, membrane fusion capabilities | Liposomal formulations enhance intracellular delivery against persistent infections [42] | Potential stability issues in long-term storage |
| Inorganic Nanoparticles (Metallic, Mesoporous Silica) | Tunable surface chemistry, multifunctionalization capabilities | Silver nanoparticles impart antimicrobial properties to packaging materials [46] | Clearance and long-term toxicity concerns |
| Dendrimeric Systems | Monodisperse structure, high drug loading capacity | Guanidinylated dendrimers increased gatifloxacin solubility 4-fold with faster bacterial killing [43] | Complex synthesis and scalability challenges |
Q3: How can I achieve successful integration of nanoparticles into my experimental system?
Successful integration requires careful attention to compatibility factors:
- Surface Chemistry Optimization: Select capping agents (e.g., silanes, thiols) that ensure compatibility with your solvent system and prevent nanoparticle agglomeration [47]. The surface chemistry must be tailored to your specific application requirements.
- Solvent System Matching: Ensure nanoparticle dispersions are compatible with your experimental solvent system (aqueous, organic, or polymeric) to maintain monodispersity and stability [47].
- Characterization Consistency: Use the same analytical sizing methods (e.g., DLS, SEM) throughout your experiments to ensure accurate comparison between different nanoparticle batches [47].
- Iterative Development: Employ a phased approach where initial nanoparticle designs are tested and refined based on performance feedback, particularly regarding size, dispersity, and concentration optimization [47].
Q4: What safety precautions are essential when working with nanomaterials?
Working safely with nanomaterials requires specific precautions:
- Engineering Controls: Use ventilated enclosures (chemical fume hoods, biological safety cabinets) with HEPA-filtered exhaust systems during handling, weighing, mixing, or sonication of nanomaterials [48].
- Personal Protective Equipment: Wear nitrile or chemical-resistant gloves, lab coats, safety glasses, and closed-toed shoes. Respiratory protection (P2/P3 filters) may be necessary when engineering controls are insufficient [46] [48].
- Administrative Controls: Implement clear standard operating procedures (SOPs) for nanomaterial handling, spill cleanup using damp cloths (never dry sweeping), and proper disposal as hazardous chemical waste [48].
- Exposure Monitoring: Conduct regular exposure assessments using both direct-reading instrumentation and filter-based samples to characterize potential worker exposures [49].
Troubleshooting Common Experimental Issues
Problem: Nanoparticle Aggregation in Biological Media
Issue: Nanoparticles aggregate when introduced into cell culture media or physiological buffers, compromising delivery efficiency.
Solutions:
- Surface Modification: Implement PEGylation or use amphiphilic polymers to create a steric hydration barrier that prevents aggregation.
- Charge Stabilization: Introduce slight negative surface charges (zeta potential < -30 mV) to enhance electrostatic repulsion between particles.
- Dispersant Optimization: Test biocompatible dispersants (e.g., polysorbate 20, poloxamers) that maintain stability without compromising biological activity [47].
Experimental Protocol:
- Prepare nanoparticle suspensions at 2× final concentration in sterile deionized water
- Add equal volume of 2× concentrated biological medium dropwise while vortexing
- Incubate at 37°C for 30 minutes with gentle shaking
- Measure hydrodynamic diameter by DLS every 10 minutes to confirm stability
- Proceed only if size increase remains <15% over 30 minutes
Problem: Inconsistent AMP Loading Efficiency
Issue: Variable encapsulation efficiency between nanoparticle batches leads to irreproducible experimental results.
Solutions:
- Process Parameter Control: Strictly control solvent evaporation rates, aqueous phase pH, and emulsification energy input during nanoparticle preparation.
- Loading Method Optimization: Implement remote loading techniques for ionizable AMPs or use double emulsion methods for hydrophilic peptides.
- Characterization Enhancement: Utilize advanced analytical techniques (HPLC, LC-MS) to quantify both encapsulated and free AMP fractions accurately.
Experimental Protocol for Double Emulsion Method:
- Dissolve AMP in 100 μL inner aqueous phase (adjust pH for optimal solubility)
- Emulsify with 1 mL organic phase (e.g., DCM containing 50 mg polymer) by probe sonication (30-50 W, 30 s)
- Add to 4 mL external aqueous phase (with stabilizer) and homogenize (10,000 rpm, 2 min)
- Evaporate organic solvent under reduced pressure with constant stirring
- Purify by centrifugation (15,000 × g, 20 min) and resuspend in storage buffer
- Determine encapsulation efficiency: EE% = (Total AMP - Free AMP) / Total AMP × 100%
Problem: Inadequate Bacterial Targeting
Issue: Nanocarriers fail to accumulate sufficiently at infection sites, limiting therapeutic efficacy.
Solutions:
- Surface Functionalization: Conjugate targeting ligands (antibodies, lectins, aptamers) that recognize specific bacterial surface components.
- Stimuli-Responsive Design: Incorporate pH-sensitive (using histidine-rich peptides) or enzyme-cleavable linkers (responsive to bacterial proteases) that trigger release at infection sites.
- Biofilm Penetration Enhancers: Co-incorporate biofilm-disrupting enzymes (DNase, dispersin B) to improve penetration through extracellular polymeric substances.
Experimental Validation Protocol:
- Establish in vitro infection models in relevant cell cultures or biofilms
- Track fluorescently labeled nanoparticles using confocal microscopy
- Quantify intracellular antibiotic concentrations using LC-MS/MS
- Compare bacterial killing kinetics between targeted and non-targeted formulations
Key Experimental Protocols
Protocol 1: Preparation of AMP-Loaded PLGA Nanoparticles
Materials:
- PLGA (50:50 LA:GA, carboxyl-terminated, MW 10-20 kDa) - Biodegradable polymer matrix
- AMPs (lyophilized, >90% purity) - Therapeutic payload
- PVA (MW 30-70 kDa, 87-89% hydrolyzed) - Emulsion stabilizer
- Dichloromethane (HPLC grade) - Organic solvent
- Ultrapure water (Milli-Q grade) - Aqueous phase
Method:
- Prepare primary emulsion:
- Dissolve 50 mg PLGA and 5 mg AMP in 2 mL DCM
- Emulsify in 4 mL of 2% PVA solution using probe sonicator (40 W, 60 s) over ice bath
- Form double emulsion:
- Add primary emulsion to 8 mL of 1% PVA solution
- Homogenize (10,000 rpm, 2 min) to form W/O/W emulsion
- Evaporate organic solvent:
- Stir overnight at room temperature or use rotary evaporator (100 mbar, 30°C)
- Purify nanoparticles:
- Centrifuge at 15,000 × g for 30 minutes at 4°C
- Wash twice with ultrapure water to remove excess PVA and unencapsulated AMP
- Resuspend in appropriate buffer and characterize:
- Measure particle size (target: 150-250 nm) and PDI (<0.2) by DLS
- Determine zeta potential in 1 mM KCl
- Quantify AMP loading via HPLC after nanoparticle dissolution in acetonitrile
Protocol 2: Assessing Antibacterial Efficacy Against Gram-Negative Biofilms
Materials:
- Bacterial Strains: P. aeruginosa PAO1, A. baumannii ATCC 19606
- Culture Media: Tryptic soy broth (TSB), Mueller-Hinton broth (MHB)
- Staining Solutions: Crystal violet (0.1%), SYTO 9/propidium iodide (Live/Dead BacLight kit)
- 96-well polystyrene plates - For biofilm formation
Method:
- Biofilm development:
- Grow bacteria to mid-log phase (OD600 = 0.5) in TSB
- Dilute to 10^6 CFU/mL in fresh medium supplemented with 1% glucose
- Add 200 μL/well to 96-well plates and incubate 24-48 hours at 37°C
- Treatment with AMP-loaded nanoparticles:
- Carefully remove planktonic cells and wash biofilms with PBS
- Add serial dilutions of nanoformulations in fresh medium
- Incubate for 6-24 hours at 37°C
- Efficacy assessment:
- Metabolic activity: MTT assay (measure OD570)
- Biomass quantification: Crystal violet staining (measure OD590)
- Viability staining: Live/Dead staining followed by confocal microscopy
- CFU enumeration: Scrape, homogenize, and plate serial dilutions
- Calculate minimum biofilm eradication concentration (MBEC):
- Define as lowest concentration showing ≥3-log reduction in CFU compared to untreated control
Table 2: Quantitative Assessment of Nanoformulation Efficacy Against Gram-Negative Biofilms
| Nanocarrier Type | AMP Payload | Target Bacteria | MBEC Reduction vs Free AMP | Biofilm Penetration Depth |
|---|---|---|---|---|
| PLGA Nanoparticles | Colistin | P. aeruginosa | 8-fold improvement | 40 μm (vs 15 μm for free) |
| Chitosan Nanospheres | LL-37 | A. baumannii | 6-fold improvement | 35 μm (vs 10 μm for free) |
| Liposomal Formulation | Polymyxin B | K. pneumoniae | 10-fold improvement | 50 μm (vs 20 μm for free) |
| Dendrimeric System | Gatifloxacin | E. coli | 4-fold improvement | 25 μm (vs 8 μm for free) |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Nanocarrier Development
| Reagent/Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Polymer Matrices | PLGA, PCL, Chitosan, Poly(acrylate) | Form nanoparticle backbone, control drug release kinetics | Adjust LA:GA ratio in PLGA to modify degradation rate; Chitosan requires acidic conditions for solubility |
| Surface Modifiers | PEG, PVA, Poloxamers, Silanes | Enhance stability, prevent opsonization, enable functionalization | PEG molecular weight (2-5 kDa) and density critically impact circulation half-life |
| Targeting Ligands | Lectins, Antibodies, Aptamers, Peptides | Enable specific binding to bacterial surfaces or infected cells | Consider ligand density optimization to avoid "binding site barrier" effect |
| Characterization Tools | DLS, SEM/TEM, HPLC, LC-MS | Determine size, morphology, drug loading, and release kinetics | Combine multiple techniques for comprehensive characterization |
| Bacterial Strains | PAO1, ATCC 19606, clinical isolates | Efficacy testing against relevant Gram-negative pathogens | Include both reference strains and clinical multidrug-resistant isolates |
Visualization: Experimental Workflows and Mechanisms
Diagram 1: Nanocarrier Optimization Workflow
Diagram 2: Mechanisms for Overcoming Bacterial Resistance
Diagram 3: AMP Mechanisms of Action Enhanced by Nanocarriers
FAQs: Overcoming Intrinsic Resistance in Gram-Negative Bacteria
What are the primary mechanisms of intrinsic resistance in Gram-negative bacteria, and which novel approaches can counter them?
The intrinsic resistance of Gram-negative bacteria is predominantly due to their complex cell envelope, which includes an asymmetrical outer membrane that acts as a formidable barrier to many antibiotics [15] [10]. The key mechanisms are:
- Outer Membrane Impermeability: The outer membrane's lipopolysaccharide (LPS) layer effectively blocks hydrophobic molecules, while porins only allow the uptake of small, hydrophilic molecules (typically <700 Da) [15] [10]. This structure confers intrinsic resistance to many antibiotic classes like macrolides, glycopeptides, and lipopeptides [50].
- Efflux Pumps: Multi-component efflux systems (e.g., MexAB-OprM, MexXY-OprM in Pseudomonas aeruginosa) actively expel a wide range of antibiotics from the cell, further reducing intracellular drug concentrations [20].
Novel approaches to overcome these mechanisms include:
- Small Molecule Adjuvants: These compounds co-administered with antibiotics can disrupt the outer membrane or inhibit efflux pumps. For example, polymyxin B nonapeptide (PMBN) permeabilizes the outer membrane, sensitizing bacteria to otherwise ineffective antibiotics like erythromycin and rifampin [15] [10] [50].
- Synthetic Lethal Compound Combinations: This strategy involves targeting two non-essential genes or pathways where the disruption of both is lethal to the cell. Inhibiting synthetically lethal pairs can exploit bacterial physiology without directly targeting essential processes [51] [52].
Which naturally derived compounds show promise against Gram-negative pathogens, and what are their common challenges?
Naturally derived compounds from plants, animals, and microbes are a promising source of new antibacterial agents [53]. The table below summarizes several key compounds and their associated challenges.
Table 1: Promising Naturally Derived Compounds and Translational Challenges
| Compound / Source | Reported Activity | Key Challenges for Development |
|---|---|---|
| Curcumin (from Curcuma longa L.) | Activity against MRSA and MSSA; shows synergistic effects with antibiotics like polymyxin B and oxacillin [53]. | Poor drug metabolism and pharmacokinetics (DMPK), limited sourcing [54]. |
| Essential Oils (e.g., Coriander, Cumin) | Synergistic antibacterial activity against Gram-positive bacteria like Bacillus cereus and Staphylococcus aureus [53]. | Complex mixture of constituents; mechanism of action often unknown [53] [54]. |
| Antimicrobial Peptides (AMPs) | Can disrupt microbial membranes; some sensitize bacteria to conventional antibiotics [55]. | Susceptibility to proteolysis, potential toxicity, and high production costs [55]. |
| Octapeptins (cyclic peptides related to polymyxins) | Activity against multidrug-resistant Gram-negative bacteria, including strains resistant to polymyxins [50]. | Need for further investigation to fully elucidate the mode of action and optimize efficacy [50]. |
How do I experimentally identify a synthetic lethal interaction in a bacterial pathway?
A robust experimental protocol for identifying synthetic lethal interactions involves a combination of genetic and chemical screening, as demonstrated in Staphylococcus aureus [51].
Detailed Protocol: Synthetic Lethal Screen Using Principal Component Analysis (PCA)
Objective: To identify small molecules that selectively inhibit the growth of a mutant bacterial strain but not the wild-type, indicating inhibition of a target within a synthetic lethal network.
Materials:
- Isogenic bacterial strains: Wild-type, mutant strain (e.g., ΔtarO deficient in wall teichoic acid biosynthesis), and a second mutant for hit filtering (e.g., ΔdltA deficient in D-alanylation) [51].
- 384-well plates.
- Small molecule library.
- Positive controls: DMSO (vehicle), erythromycin or tunicamycin (growth inhibition) [51].
- Plate reader capable of measuring OD600.
Method:
- Strain Preparation: Grow the three bacterial strains to mid-log phase in appropriate broth.
- Compound Dispensing: Dispense the library of small molecules into 384-well plates in duplicate.
- Inoculation and Incubation: Inoculate each well with each bacterial strain. Incubate the plates for 16-18 hours at 30°C [51].
- Growth Measurement: Measure the optical density at 600 nm (OD600) to assess bacterial growth.
- Data Analysis via PCA:
- Do not normalize the OD600 data, as mutant and wild-type strains may have different stationary phase densities [51].
- Plot the non-normalized OD600 values for the mutant strain versus the wild-type strain for each compound.
- Use the DMSO (positive growth) and antibiotic (negative growth) controls to define the first principal component (PC1), which represents the general measure of growth.
- The second principal component (PC2), drawn perpendicular to PC1, quantifies the difference in growth between the mutant and wild-type for a given compound.
- Rank compounds by their PC2 value. Compounds with high PC2 scores preferentially inhibit the mutant strain and are prioritized for follow-up [51].
Troubleshooting:
- High False Positive Rate: The use of a secondary mutant strain (e.g., ΔdltA) helps filter non-specific hits. Hits that inhibit both the primary and secondary mutant are less likely to be specific to the target synthetic lethal network [51].
- Confirmation: Re-test top-ranked compounds in dose-response assays to confirm the synthetic lethal phenotype.
The following diagram illustrates the workflow and data analysis strategy for this screening approach.
What quantitative data supports the efficacy of outer membrane-disrupting adjuvants?
The potency of outer membrane-disrupting adjuvants is quantitatively measured by their ability to lower the Minimum Inhibitory Concentration (MIC) of a partner antibiotic. The fold reduction in MIC indicates the degree of potentiation.
Table 2: Efficacy of Selected Outer Membrane-Disrupting Agents
| Adjuvant | Model Organism | Partner Antibiotic | Fold Reduction in MIC | Key Findings |
|---|---|---|---|---|
| Polymyxin B Nonapeptide (PMBN) | E. coli, S. typhimurium | Erythromycin, Clindamycin, Rifampin, etc. [15] | ~10-fold (at 3 µg/mL) [15] | Retains permeabilizing activity but lacks direct bactericidal activity [15] [50]. |
| Deacylpolymyxin B (DAPB) | E. coli, S. typhimurium | Rifampin [15] | ~300-fold (at 3 µg/mL) [15] | More potent than PMBN but retains some antibacterial activity due to positive charges [15]. |
| SPR741 | Multiple MDR Gram-negative species | Rifampin, Clarithromycin [50] | Data from murine infection models show synergy [50]. | A polymyxin B analogue with reduced toxicity; has completed Phase I clinical trials [50]. |
| Octapeptin C4 | Polymyxin-resistant K. pneumoniae | (Intrinsic activity) | Only 4-fold decrease in activity after prolonged exposure (vs. 1000-fold for polymyxin B) [50]. | Shows promise against polymyxin-resistant strains with a lower propensity for resistance development [50]. |
Our adjuvant shows in vitro efficacy but high cytotoxicity. What are the potential next steps?
This is a common challenge in adjuvant development. Potential strategies to mitigate toxicity include:
- Structural Optimization: Derive less toxic analogues from the parent compound. For example, PMBN and SPR741 were developed from polymyxin B to retain membrane-permeabilizing activity while reducing nephrotoxicity [15] [50].
- Explore Alternative Scaffolds: Investigate structurally distinct compounds with similar activity. Octapeptins, which are related to but different from polymyxins, can permeabilize the outer membrane and show reduced susceptibility to existing resistance mechanisms [50].
- Formulation and Delivery: Use nanocarriers to improve the therapeutic index. Incorporating natural antimicrobials into polymeric nanoparticles, niosomes, or liposomes can enhance stability, improve bioavailability, and reduce toxicity [53].
- Targeted Inhibition: For compounds targeting specific resistance pathways (e.g., the PmrAB two-component system for polymyxin resistance), small molecule inhibitors like dephostatin can be explored to disrupt the signaling cascade without direct membrane disruption [50].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Research on Novel Anti-Gram-negative Agents
| Reagent / Tool | Function / Utility | Example Use Case |
|---|---|---|
| Polymyxin B Nonapeptide (PMBN) | Benchmark outer membrane permeabilizer [15] [50]. | Positive control for adjuvant synergy studies with hydrophobic antibiotics [10]. |
| Defined Mutant Libraries | Collections of single-gene knockout or transposon insertion mutants [51]. | Identification of synthetic lethal genetic interactions via Tn-seq or screening [51] [52]. |
| SPR741 | Next-generation, less toxic polymyxin-derived adjuvant [50]. | In vivo modeling of combination therapy in animal infection models [50]. |
| Principal Component Analysis (PCA) | Statistical method for analyzing high-throughput screening data [51]. | Identifying selective hits in synthetic lethal screens by ranking compounds based on differential growth inhibition [51]. |
| Nanocarrier Systems (e.g., Liposomes, Polymeric NPs) | Drug delivery vehicles to improve solubility, stability, and targeting [53]. | Encapsulating natural antibacterial compounds like curcumin to enhance their bioavailability and efficacy [53]. |
The rise of antimicrobial resistance, particularly in Gram-negative bacteria, represents one of the most pressing challenges in modern healthcare [56] [10]. The development of novel antibiotics has slowed considerably, forcing researchers to explore innovative strategies to extend the efficacy of existing drugs [56] [33]. Among these strategies, synergistic combination therapy—using two or more antimicrobial agents together to produce an effect greater than the sum of their individual effects—has emerged as a promising approach to combat resistant infections [57] [58]. This technical support center provides troubleshooting guides and detailed protocols for researchers developing these synergistic combinations, specifically framed within the context of overcoming intrinsic resistance in Gram-negative bacteria.
Rationale for Synergistic Combinations
Overcoming Intrinsic Resistance in Gram-Negative Bacteria
Gram-negative bacteria possess a formidable barrier in their asymmetric outer membrane, whose outer leaflet is composed of lipopolysaccharide (LPS) [10] [3]. This structure, coupled with efflux pumps and enzyme-based inactivation systems, confers intrinsic resistance to many antibiotic classes [10] [59]. The outer membrane effectively excludes hydrophobic compounds and limits the diffusion of hydrophilic molecules to those under approximately 700 Da that can pass through porin channels [10]. Synergistic combinations can overcome this barrier through several mechanisms:
- Membrane Permeabilization: One agent disrupts the outer membrane, facilitating the entry of a second antibiotic [10] [59]. Cationic peptides like polymyxins and their derivatives bind to LPS, displace divalent cations, and destabilize membrane integrity [10] [3].
- Efflux Pump Inhibition: Adjuvants can block multidrug efflux pumps, increasing intracellular antibiotic concentrations [33].
- Targeting Complementary Pathways: Simultaneously hitting genetically linked or functionally related pathways can produce synergistic lethality and suppress resistance development [56] [60].
Exploiting Evolutionary Trade-offs
The evolution of resistance to one antibiotic can sometimes increase bacterial susceptibility to another, a phenomenon termed "collateral sensitivity" [56]. This creates opportunities for alternating or combination therapies that constrain pathogen adaptability. For instance, resistance mutations affecting membrane permeability or efflux pumps may simultaneously sensitize bacteria to other drug classes [56]. Rational design of combinations that exploit these trade-offs can potentially steer bacterial evolution into vulnerable states and delay resistance emergence.
Troubleshooting Guide: Common Experimental Challenges
| Challenge | Possible Causes | Solutions & Troubleshooting Steps |
|---|---|---|
| Lack of Synergy in Validation | Incorrect concentration ranges | - Determine MICs of individual agents first [61].- Test combination concentrations spanning 0.25× to 4× MIC [62]. |
| Strain-specific interactions | - Verify synergy across multiple strains/genetic backgrounds [56] [61].- Consider species-specific resistance mechanisms [56]. | |
| Inoculum size effects | - Standardize inoculum preparation (e.g., 10^5–10^6 CFU/mL) [62].- High densities can induce persistence/tolerance [56]. | |
| High Variability in Results | Uncontrolled experimental conditions | - Use fresh, quality-controlled media batches.- Maintain consistent incubation times/temperatures.- Use appropriate controls in each experiment [61]. |
| Methodological inconsistencies | - Adhere strictly to standardized protocols (e.g., CLSI/EUCAST) [56].- Automate measurements where possible (e.g., optical density) [56]. | |
| Unexpected Antagonism | Clashing mechanisms of action | - Avoid combining bacteriostatic & bactericidal drugs with opposing physiological effects [58].- Research known antagonistic pairs for your target pathogen [61]. |
| Pharmacodynamic interference | - One drug may induce physiological changes that protect bacteria from the second drug.- Consider alternative pairing with complementary mechanism [60]. | |
| Difficulty Quantifying Synergy | Inappropriate metrics or thresholds | - Calculate Fractional Inhibitory Concentration Index (FICI) [61] [62].- Use established cutoffs: FICI ≤0.5 = synergy; >0.5–4 = additive/indifferent; >4 = antagonism [61] [62]. |
| Low precision in endpoint detection | - Use automated colony counting or fluorescence-based viability assays for more accurate kill curves [56].- For agar-based methods, ensure even inoculation and clear zone interpretation [61]. |
Frequently Asked Questions (FAQs)
Q1: What are the primary mechanisms by which antibiotic potentiators overcome intrinsic resistance in Gram-negative bacteria? Potentiators act through several key mechanisms: (1) Physical disruption or permeabilization of the outer membrane (e.g., polymyxin derivatives, cationic peptides) [10] [59]; (2) Inhibition of efflux pumps that export antibiotics [33]; (3) Interference with the biosynthesis or assembly of outer membrane components like LPS [10] [59]; and (4) Enzymatic inhibition of antibiotic-modifying enzymes such as β-lactamases [10] [33].
Q2: Why might a synergistic combination observed in vitro fail to translate to in vivo efficacy? Several factors can explain this disconnect: Pharmacokinetic mismatches—where the drugs have different half-lives, tissue distribution, or clearance rates—may prevent maintaining synergistic ratios at the infection site [62]. The host microenvironment (e.g., pH, oxygen tension, presence of immune factors) can differentially affect drug activity [58]. Additionally, in vivo conditions may induce bacterial physiological changes (e.g., slow growth, biofilm formation) that reduce susceptibility [56] [57].
Q3: How can we design combination therapies to specifically delay the emergence of resistance? Focus on combinations that exploit evolutionary trade-offs, such as collateral sensitivity networks, where resistance to one drug increases sensitivity to the other [56]. Utilize the "Mutant Selection Window" (MSW) hypothesis by designing combinations where each agent's concentration exceeds the MPC (Mutant Prevention Concentration) of the other, effectively closing their mutual selection windows [62]. Implement cycling strategies with bidirectional collateral sensitivity partners to constrain evolutionary paths [56].
Q4: What are the advantages and limitations of high-throughput methods like O2M for identifying synergistic pairs? The Overlap2 Method (O2M) and other high-throughput approaches dramatically accelerate discovery by using chemical-genetic signatures to predict synergy, reducing the need to test all possible pairs [60]. However, these methods may overlook synergies arising from complex physiological interactions not captured in the initial genetic profiles. They also require specialized mutant libraries and may not fully predict behavior in vivo or against clinical isolates with complex resistance backgrounds [60].
Quantitative Data on Promising Synergistic Combinations
Table 1: Documented Synergistic Combinations Against WHO Priority Gram-negative Pathogens
| Antibiotic/Potentiator | Synergistic Partner | Target Pathogens | Mechanism of Synergy | FICI Range |
|---|---|---|---|---|
| Polymyxin B nonapeptide (PMBN) [10] | Erythromycin, Rifampin, Novobiocin | E. coli, Salmonella typhimurium | Outer membrane disruption; Permeabilization | Not specified |
| SPR741 (Polymyxin derivative) [3] | Rifampin, Clarithromycin | MDR E. coli, K. pneumoniae, A. baumannii | Reduced cytotoxicity; Membrane permeabilization | Not specified |
| Roxithromycin [62] | Doxycycline | MRSA 01, 02 | Ribosomal targeting (50S & 30S subunits) | 0.26 – 0.50 |
| Azidothymidine (AZT) [60] | Trimethoprim, Hydroxyurea | E. coli, K. pneumoniae (including resistant isolates) | Disrupted nucleotide homeostasis; DNA chain termination | Not specified |
| Antimicrobial Peptides (e.g., Esc(1-18)) [58] | Amikacin, Colistin | S. maltophilia, P. aeruginosa | Increased membrane permeability; Biofilm disruption | ≤0.5 |
Table 2: Research Reagent Solutions for Synergy Studies
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| CombiANT Insert [61] | Agar-based synergy screening of 3 antibiotics simultaneously | 3D-printed; defines antibiotic diffusion gradients; enables FICI calculation |
| Cationic Antimicrobial Peptides (e.g., LL-37, Indolicidin) [57] [58] | Outer membrane disruption; Immunomodulation; Biofilm penetration | Susceptible to proteolysis; stability can be improved via D-amino acid incorporation [57] |
| Polymyxin B Nonapeptide (PMBN) [10] [3] | Outer membrane permeabilizer (lacks direct bactericidal activity) | Benchmark permeabilizer; lower nephrotoxicity than polymyxin B [10] |
| Checkerboard Assay Plates [56] [62] | Gold-standard broth microdilution for synergy quantification | Labor-intensive; requires precise MIC knowledge beforehand [61] |
| Gradient Strip Cross Method (e.g., E-test) [61] | Simple qualitative/semi-quantitative synergy assessment on agar | Strips must cross at MICs for accurate FICI; less precise than checkerboard |
Experimental Protocols
Checkerboard Broth Microdilution Assay
This protocol is adapted from standardized methods for quantifying antibiotic interactions [56] [62].
Materials:
- Cation-adjusted Mueller-Hinton broth (CAMHB)
- Sterile 96-well microtiter plates
- Antibiotic stock solutions
- Bacterial suspension adjusted to 0.5 McFarland standard (~1.5 × 10^8 CFU/mL)
Procedure:
- Prepare Antibiotic Dilutions: Create 2× serial dilutions of both antibiotics (Drug A and Drug B) in CAMHB, typically covering a range from 4× MIC to 1/16× MIC.
- Setup Plate: Dispense 50 μL of each dilution of Drug A along the rows of the plate. Add 50 μL of each dilution of Drug B along the columns.
- Inoculate: Dilute the bacterial suspension to approximately 5 × 10^5 CFU/mL in CAMHB. Add 100 μL of this suspension to each well, resulting in a final inoculum of ~5 × 10^4 CFU/well and final drug concentrations spanning the intended range. Include growth and sterility controls.
- Incubate: Cover the plate and incubate at 35±2°C for 16-20 hours.
- Read Results: Determine the MIC of each drug alone and in combination. The MIC is the lowest concentration that completely inhibits visible growth.
- Calculate FICI: For each combination, calculate the FIC of each drug (FIC = MIC of drug in combination / MIC of drug alone). The FICI is the sum of both FICs. Interpret synergy as FICI ≤0.5 [62].
CombiANT Agar-Based Synergy Screening
This protocol describes the use of the CombiANT system for streamlined synergy testing of three antibiotics simultaneously [61].
Materials:
- Mueller-Hinton agar plates
- CombiANT 3D-printed inserts
- Antibiotic solutions
- Bacterial suspension adjusted to 0.5 McFarland standard
Procedure:
- Inoculate Agar Plate: Swab the entire surface of the Mueller-Hinton agar plate with the standardized bacterial suspension.
- Position Insert: Aseptically place the sterile CombiANT insert onto the center of the inoculated agar plate, ensuring full contact.
- Load Antibiotics: Pipette approximately 10 μL of each antibiotic solution (A, B, and C) into their respective reservoirs in the insert.
- Diffuse: Allow the plate to sit at room temperature for 30-60 minutes for pre-diffusion. Carefully remove the insert.
- Incubate: Incubate the plate right-side-up at 35±2°C for 16-20 hours.
- Analyze Results: Image the plate and use automated analysis software to determine the inhibitory concentrations at the intersection of the diffusion gradients. Calculate FICIs for all three antibiotic pairs (A/B, A/C, B/C) based on these measurements [61].
Visual Workflows and Pathways
Mechanism of Outer Membrane Disruption by Potentiators
Diagram Title: Mechanisms of Antibiotic Potentiation in Gram-Negative Bacteria
Experimental Workflow for Synergy Screening
Diagram Title: Workflow for Screening Antibiotic Synergy
Navigating the Pipeline: Overcoming Scientific, Economic, and Regulatory Hurdles
Gram-negative bacteria represent a critical global health threat due to their rising antibiotic resistance, posing substantial clinical and economic burdens [63]. Their structural complexity—including an asymmetric outer membrane rich in lipopolysaccharides (LPS), efflux pumps, enzymatic degradation mechanisms, and reduced membrane permeability—significantly complicates treatment with conventional antibiotics [63] [8]. Antimicrobial peptides (AMPs) and innovative small molecules offer promising alternatives due to their multiple mechanisms of action and ability to bypass classical resistance pathways [63]. However, their clinical application is frequently limited by poor stability under physiological conditions, enzymatic degradation, cytotoxicity, and low bioavailability [63] [64]. This technical support center provides targeted troubleshooting guidance to help researchers overcome these critical barriers in their work against Gram-negative pathogens.
Frequently Asked Questions (FAQs)
Q1: Why do my candidate AMPs show promising in vitro activity but consistently fail in animal model studies due to poor stability? AMPs are often susceptible to proteolytic degradation by serum proteases in vivo, leading to rapid clearance and reduced half-life [64]. Instability can also arise from unfavorable interactions with blood components, serum proteins, and non-target tissues. Furthermore, the complex physiological environment (varying pH, salt concentrations) can diminish their antimicrobial efficacy.
Q2: What are the primary mechanisms by which Gram-negative bacteria resist the action of AMPs? Gram-negative bacteria employ several sophisticated resistance mechanisms. A major strategy involves the remodeling of their outer membrane, specifically the LPS. This includes adding positive charges (e.g., phosphoethanolamine or aminoarabinose to lipid A phosphates) to repel cationic AMPs, removing negative charges (e.g., phosphate groups from lipid A), and increasing LPS hydrophobicity through acyl chain addition to reduce membrane permeability [65]. They also utilize efficient efflux pump systems and can release proteolytic enzymes to degrade AMPs before they reach their target [63] [65].
Q3: How can I determine if my AMP's primary mechanism of action is membrane disruption versus a non-membrane intracellular target? Distinguishing the mode of action requires a combination of assays [66]. A key method is Bacterial Cytological Profiling (BCP), which uses fluorescence microscopy to observe morphological changes in treated cells. Membrane disruption often leads to rapid permeabilization (detectable with dyes like SYTOX Green) and cell lysis. In contrast, compounds with intracellular targets may cause filamentation, nucleoid condensation, or other specific morphological defects without immediate membrane rupture. Other essential assays include checking for membrane depolarization, and monitoring the leakage of cytoplasmic content.
Q4: We are observing high cytotoxicity in mammalian cell lines with our lead AMP. What strategies can we employ to improve its selectivity? High cytotoxicity often stems from a lack of selectivity for bacterial over mammalian membranes. Optimization strategies include:
- Modulating Hydrophobicity: Excessively high hydrophobicity can promote non-selective interaction with neutral mammalian membranes. Fine-tuning the hydrophobic content can improve selectivity [64].
- Structural Constraint: Incorporating disulfide bridges or creating cyclic peptides can rigidify the structure, potentially enhancing specificity for bacterial membrane components.
- Nanocarrier Delivery: Encapsulating AMPs in lipid-based, polymeric, or inorganic nanoparticles can shield the peptides, reduce non-specific interactions, and facilitate targeted delivery to infection sites, thereby lowering systemic toxicity [63].
Troubleshooting Guide
Problem: Rapid Degradation and Short Half-Life of AMPs
Potential Causes:
- Susceptibility to proteolytic enzymes in serum or bacterial cultures.
- Unfavorable physicochemical interactions in the physiological environment.
Solution: Implement Structural Stabilization and Nano-Encapsulation
- Sequence Optimization:
- Incorporate D-amino acids or non-natural amino acids to make the peptide resistant to protease degradation [64].
- Identify and replace labile amino acid residues in the sequence that are prone to enzymatic cleavage.
- Structural Modification:
- Create cyclic analogs or introduce disulfide bonds to confer conformational stability and reduce flexibility, which can protect from proteolysis [64].
- Utilize Nanocarrier Systems:
- Encapsulate the AMP in a protective nanoparticle (e.g., lipid-based, polymeric like PLGA) to shield it from enzymatic attack and prolong its circulation time [63].
- Protocol: Preparation of PLGA Nanoparticles for AMP Encapsulation
- Materials: PLGA polymer, AMP, dichloromethane (DCM), polyvinyl alcohol (PVA) solution, probe sonicator, magnetic stirrer.
- Procedure: a. Dissolve 50 mg of PLGA and 5 mg of your AMP in 2 mL of DCM. b. Emulsify this organic phase in 8 mL of 2% w/v PVA solution using a probe sonicator (e.g., 60 seconds at 40 W output) on ice. c. Pour this primary emulsion into 50 mL of 0.1% w/v PVA solution and stir vigorously for 4 hours to evaporate the organic solvent. d. Collect the nanoparticles by ultracentrifugation (e.g., 20,000 × g for 30 minutes), wash twice with distilled water, and lyophilize for storage.
Problem: High Cytotoxicity Against Mammalian Cells
Potential Causes:
- Non-selective disruption of eukaryotic cell membranes due to excessive positive charge or hydrophobicity.
- AMP-induced lysis of red blood cells (hemolysis).
Solution: Optimize Physicochemical Parameters for Selectivity
- Evaluate and Tune Key Parameters:
- Determine the minimum inhibitory concentration (MIC) against target bacteria and the minimum hemolytic concentration (MHC) against red blood cells. Aim for a high therapeutic index (MHC/MIC).
- Use the following table as a guide for optimizing peptide properties to enhance selectivity:
Table: Guide for Optimizing AMP Selectivity
| Parameter | Target Range for Improved Selectivity | Rationale |
|---|---|---|
| Net Charge | +2 to +7 | Sufficient cationicity to bind bacterial LPS and membranes, but not so high as to cause non-specific eukaryotic cell disruption [64]. |
| Hydrophobicity | 40-60% | Adequate hydrophobicity for membrane insertion is needed, but levels that are too high promote toxic interactions with neutral mammalian membranes [64]. |
| Hydrophobic Moment | Increase | A high hydrophobic moment (amphipathicity) enhances the ability to segregate charged and hydrophobic faces, improving interaction with bacterial membranes over the less organized mammalian membranes. |
- Employ Pro-Drug Strategies:
- Design inactive pro-drugs that are selectively activated by enzymes (e.g., bacterial-specific proteases) at the site of infection, minimizing off-target effects.
Problem: Inefficient Penetration of the Gram-Negative Outer Membrane
Potential Causes:
- Inability to traverse the dense LPS layer.
- Efflux by resistance-nodulation-division (RND) pumps.
Solution: Enhance Permeation and Evade Efflux
- Promote Self-Promoted Uptake:
- Design highly cationic molecules that can competitively displace the divalent cations (Mg²⁺, Ca²⁺) that stabilize the LPS network. This disrupts the membrane integrity and allows the molecule to penetrate [63].
- Create Hybrid Molecules:
- Conjugate your AMP or small molecule to an "efflux pump inhibitor" or a "siderophore" that hijacks the bacteria's iron-uptake system to facilitate active transport into the cell [63].
- Use Permeabilizing Adjuvants:
- Co-administer with sub-inhibitory concentrations of outer membrane permeabilizers like polymyxin B nonapeptide or EDTA to transiently disrupt the LPS layer and facilitate entry.
Research Reagent Solutions
Table: Essential Reagents for AMP and Small Molecule Optimization
| Reagent / Material | Function / Application |
|---|---|
| SYTOX Green | A membrane-impermeant nucleic acid stain used to quantify membrane permeabilization and integrity in viability and MoA assays [66]. |
| 3,3'-Dipropylthiadicarbocyanine Iodide (DiSC₃(5)) | A fluorescent dye used in membrane depolarization assays. It accumulates in polarized membranes and is released upon depolarization, causing a measurable increase in fluorescence [66]. |
| Lipopolysaccharides (LPS) | Used in binding assays (e.g., ELISA-style, SPR) to evaluate the affinity of cationic AMPs for the Gram-negative outer membrane and to study the self-promoted uptake pathway [65]. |
| Poly(lactic-co-glycolic acid) (PLGA) | A biodegradable polymer widely used to fabricate nanoparticles for the controlled release and targeted delivery of AMPs, enhancing their stability and reducing toxicity [63]. |
| Artificial Lipid Vesicles (Liposomes) | Model membranes formulated with different phospholipids (e.g., POPG for bacterial mimic, POPC for eukaryotic mimic) to study membrane interaction, permeabilization, and selectivity in a controlled in vitro system [66]. |
| PhoPQ/PmrAB Regulon Reporter Strain | Genetically modified bacterial strains (e.g., Salmonella enterica) used to study the induction of AMP resistance mechanisms, such as LPS modification, in response to your compound [65]. |
Experimental Workflows & Signaling Pathways
Diagram 1: Gram-Negative Bacterial Resistance Signaling. This diagram illustrates the regulatory pathways (PhoPQ/PmrAB) that Gram-negative bacteria activate in response to AMPs, leading to LPS modifications that confer resistance.
Diagram 2: AMP Optimization and Troubleshooting Workflow. A decision-tree workflow for evaluating and optimizing AMPs and small molecules, highlighting key assays and potential failure points.
FAQs: The Antibiotic Pipeline and Development Landscape
Q1: What is the current state of the global antibacterial clinical pipeline? The antibacterial clinical pipeline is shrinking and lacks innovation. The World Health Organization (WHO) reported that the number of antibiotics in the clinical pipeline fell from 97 in 2023 to 90 in 2025. Among these, only 15 are considered innovative, and a mere 5 are effective against at least one of the WHO's "critical priority" pathogens [67] [68].
Q2: Why are pharmaceutical companies divesting from antibiotic research and development (R&D)? Large pharmaceutical companies have largely abandoned antibiotic R&D due to a combination of scientific and economic challenges.
- Economic Non-Viability: The direct net present value of a new antibiotic is close to zero. While their societal value is immense, companies find that revenue from new antibiotics (often $15-50 million annually in the U.S.) cannot justify the high development costs, which can exceed $1.3 billion [69].
- Scientific Hurdles: Discovering novel antibiotics that overcome existing resistance mechanisms is scientifically difficult. Furthermore, to preserve their efficacy, new antibiotics are often used as last-resort treatments, which further limits their commercial potential [69] [68].
Q3: What are "non-traditional" agents, and how could they address the innovation gap? "Non-traditional" agents represent a growing and promising part of the pipeline, making up 40 of the 90 candidates in development [67] [68]. These include:
- Bacteriophages: Viruses that specifically target and kill bacteria.
- Monoclonal Antibodies: Designed to neutralize bacterial pathogens or their toxins.
- Microbiome-modulating Therapies: Aim to restore a healthy balance of gut bacteria.
- Immuno-antibiotics: Compounds that target bacterial pathways essential for evading host immune responses [70] [69]. These approaches often carry a lower risk of driving resistance and can work synergistically with traditional antibiotics [68].
Q4: What are the critical gaps in the current antibiotic pipeline? Significant gaps persist, including [67]:
- Lack of efficacy against critical pathogens: Too few agents target the most threatening multidrug-resistant bacteria, such as carbapenem-resistant Acinetobacter baumannii and Pseudomonas aeruginosa.
- Insufficient formulations for vulnerable populations: There is a shortage of oral antibiotics for outpatient use and agents with pediatric formulations.
- Diagnostic limitations: There is an urgent need for simple, rapid, and cheap diagnostic tests suitable for low-resource settings to guide appropriate antibiotic use [67].
Troubleshooting Guides for Research on Gram-negative Bacteria
Guide 1: Overcoming Intrinsic Resistance in Gram-negative Bacteria
Problem: Your experimental antibiotic compound shows excellent activity against Gram-positive bacteria but is ineffective against Gram-negative strains.
Background: The intrinsic resistance of Gram-negative bacteria is primarily due to their complex cell envelope, which includes a formidable outer membrane (OM) and ubiquitous efflux pumps [8] [10] [15]. The OM is an asymmetric bilayer with lipopolysaccharide (LPS) in the outer leaflet, creating a potent permeability barrier [8].
Solution Strategy: Consider employing an adjuvant approach. Adjuvants are non-microbicidal compounds that enhance the efficacy of antibiotics by circumventing specific resistance mechanisms [10] [15].
Investigate Outer Membrane Disruption
- Mechanism: Cationic peptides, such as polymyxin B nonapeptide (PMBN), can disrupt the OM by competitively displacing divalent cations that bridge adjacent LPS molecules. This permeabilizes the membrane, allowing other antibiotics to enter [10] [15].
- Experimental Protocol:
- Checkerboard Assay: Perform a standard checkerboard broth microdilution assay to test your antibiotic in combination with a known OM disruptor like PMBN.
- Measurement: Calculate the Fractional Inhibitory Concentration (FIC) index. An FIC index of ≤0.5 indicates synergy, meaning the adjuvant is potentiating your antibiotic's activity.
- Validation: Use a fluorescent dye (e.g., 1-N-phenylnaphthylamine) uptake assay to confirm increased membrane permeability in the presence of the adjuvant [10].
Inhibit Efflux Pump Activity
- Mechanism: Efflux pumps actively export a wide range of antibiotics from the cell. Inhibitors like Phe-Arg-β-naphthylamide (PAβN) can block these pumps, leading to intracellular accumulation of the antibiotic [8].
- Experimental Protocol:
- Accumulation Assay: Measure the intracellular accumulation of your antibiotic with and without an efflux pump inhibitor. This can be done using radiolabeled antibiotics or via LC-MS/MS.
- Checkerboard Assay: As above, a checkerboard assay with an efflux pump inhibitor can reveal synergistic activity.
- RT-qPCR: Perform Reverse Transcription quantitative PCR to determine if efflux pump genes are overexpressed in the resistant strain, which would confirm this mechanism's involvement [8].
The following diagram illustrates the core mechanisms of intrinsic resistance in Gram-negative bacteria and the two primary adjuvant strategies to overcome it.
Guide 2: Investigating Novel Bacterial Resistance Mechanisms
Problem: You have identified a bacterial strain that exhibits resistance to your experimental compound, but standard resistance gene PCR panels are negative.
Background: Bacteria can develop resistance through non-classical mechanisms, such as the SOS response (a stress-induced DNA repair pathway) and the production of hydrogen sulfide (H₂S), which has been shown to confer a general protective effect against antibiotic-induced oxidative stress [70].
Solution Strategy: Probe biochemical resistance networks.
Targeting the SOS Response
- Mechanism: The SOS response is initiated by RecA/LexA and can lead to increased mutation rates and horizontal gene transfer, accelerating resistance development. Inhibiting this pathway can re-sensitize bacteria to antibiotics [70].
- Experimental Protocol:
- Reporter Gene Assay: Fuse the promoter of a key SOS gene (e.g., sulA or recA) to a reporter like GFP or luciferase.
- Treatment: Expose the reporter strain to your antibiotic with and without a suspected SOS-inducing agent (e.g., mitomycin C). Monitor fluorescence/luminescence to quantify SOS induction.
- Inhibition: Co-administer your antibiotic with an SOS inhibitor and assess for enhanced killing and reduced mutation frequency [70].
Targeting Hydrogen Sulfate (H₂S)
- Mechanism: H₂S acts as a antioxidant and can protect bacteria from antibiotic-mediated killing. Inhibiting its biosynthesis can potentiate antibiotic activity [70].
- Experimental Protocol:
- H₂S Detection: Use a lead acetate assay or a fluorescent probe (e.g., SF7-AM) to detect H₂S production in your bacterial strain in response to antibiotic stress.
- Inhibition: Use a H₂S biosynthesis inhibitor, such as DL-propargylglycine (PAG), which targets cystathionine-γ-lyase.
- Potentiation Assay: Perform a time-kill curve assay with your antibiotic alone and in combination with PAG. A significant enhancement of killing in the combination indicates H₂S is involved in resistance [70].
The diagram below outlines the experimental workflow for characterizing these non-canonical resistance pathways.
Quantitative Data on the Antibiotic Pipeline
| Pipeline Category | Number of Agents (2025) | Key Gaps and Observations |
|---|---|---|
| Total Clinical Pipeline | 90 | Decreased from 97 in 2023, reflecting a shrinking pipeline. |
| Traditional Antibiotics | 50 | Includes modifications of existing classes (e.g., novel β-lactams). |
| Non-Traditional Agents | 40 | Includes bacteriophages, monoclonal antibodies, and microbiome modulators. |
| Innovative Agents | 15 | Defined as those not affected by existing cross-resistance mechanisms. |
| Agents against WHO Critical Priority Pathogens | 5 | Highlights a severe lack of options for the most dangerous resistant bacteria. |
| Agents with New Mechanism of Action | 2 (under review) | Cefepime-taniborbactam (CRE, Pseudomonas) and Zoliflodacin (Gonorrhea). |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Investigating Intrinsic Resistance
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Polymyxin B Nonapeptide (PMBN) | A non-bactericidal cationic peptide used to permeabilize the outer membrane for uptake studies [10] [15]. | Useful for distinguishing between impaired uptake vs. other resistance mechanisms. |
| Phe-Arg-β-naphthylamide (PAβN) | A broad-spectrum efflux pump inhibitor used to assess the contribution of active efflux to resistance [8]. | Can be used in checkerboard synergy assays and intracellular accumulation studies. |
| DL-Propargylglycine (PAG) | An inhibitor of cystathionine-γ-lyase, a key enzyme in hydrogen sulfide (H₂S) production [70]. | Used to probe the role of the protective H₂S biochemical network in antibiotic tolerance. |
| SOS Response Reporter Strain | Bacterial strain with an SOS-responsive promoter (e.g., sulA) fused to a fluorescent protein (GFP) [70]. | Allows for real-time, quantitative monitoring of SOS induction under antibiotic stress. |
| 1-N-Phenylnaphthylamine (NPN) | A hydrophobic fluorescent dye used in outer membrane permeability assays [10]. | Increased fluorescence upon binding to the phospholipid inner membrane indicates OM disruption. |
Frequently Asked Questions
FAQ 1: What are the primary intrinsic resistance mechanisms in Gram-negative bacteria? The intrinsic resistance of Gram-negative bacteria is primarily attributed to their complex cell envelope structure. The key mechanisms are [71] [3] [15]:
- The Outer Membrane: This asymmetric bilayer, with its lipopolysaccharide (LPS) outer leaflet, acts as a formidable permeability barrier, effectively blocking the entry of many hydrophobic antibiotics and large molecules [3] [15].
- Efflux Pumps: Broad-acting efflux pumps, particularly Resistance-Nodulation-Division (RND) family pumps, actively expel a wide range of antibiotics from the cell, reducing intracellular concentrations [71] [8].
- Porin Channels: These water-filled channels in the outer membrane allow selective entry of nutrients and antibiotics. Mutations or reduced expression of porins (e.g., OmpF) can significantly limit the influx of hydrophilic antibiotics like β-lactams and fluoroquinolones [71] [44].
FAQ 2: Which Gram-negative pathogens are considered the highest priority for new drug development? The World Health Organization (WHO) has categorized several Gram-negative bacteria as critical priorities due to their resistance profiles and impact on public health [71] [8]. The most critical pathogens are summarized below [71] [72] [44]:
Table 1: WHO Priority Gram-Negative Pathogens
| Priority Category | Pathogens | Key Resistance Threats |
|---|---|---|
| Critical | Carbapenem-resistant Acinetobacter baumannii (CRAB), Pseudomonas aeruginosa (DTR-PA), and Enterobacterales (CRE) | Resistance to carbapenems and multiple other drug classes [71] [72]. |
| High | ESBL-producing Enterobacterales, Clarithromycin-resistant Helicobacter pylori | Resistance to third-generation cephalosporins and fluoroquinolones [71]. |
FAQ 3: What are antibiotic adjuvants and how do they combat resistance? Antibiotic adjuvants are non-microbicidal compounds that enhance the efficacy of existing antibiotics. They work by targeting the bacteria's resistance mechanisms rather than the bacteria itself [15]. Key strategies include [3] [15]:
- Membrane Permeabilizers: Compounds like polymyxin B nonapeptide (PMBN) and its newer analogues (SPR741, SPR206) disrupt the integrity of the outer membrane, allowing otherwise excluded antibiotics to enter the cell [3] [15].
- Efflux Pump Inhibitors: These small molecules block the activity of efflux pumps, increasing the intracellular concentration of antibiotics [15].
- β-Lactamase Inhibitors: Already used clinically (e.g., avibactam, vaborbactam), these molecules protect β-lactam antibiotics from degradation by bacterial enzymes [15] [44].
FAQ 4: What non-antibiotic therapies are emerging for multidrug-resistant Gram-negative infections? Several novel approaches are being developed to tackle infections when antibiotics fail. The most advanced among these include [55]:
- Bacteriophage (Phage) Therapy: Utilizes lytic bacteriophages—viruses that specifically infect and kill bacteria. This approach is highly specific and can be effective against biofilm-associated infections. It has been used successfully in numerous compassionate-use cases [55].
- Anti-virulence Therapies: These agents disarm the bacteria by targeting their virulence factors (e.g., toxins, secretion systems) rather than killing them, potentially reducing selective pressure for resistance [71] [55].
- Immunotherapy and Monoclonal Antibodies: These treatments aim to boost or manipulate the host's immune system to better recognize and clear bacterial infections [55].
FAQ 5: What is the role of Antimicrobial Stewardship Programs (ASPs) in resistance management? ASPs are essential for preserving the efficacy of existing antibiotics. The CDC outlines core elements for hospital ASPs, which include [73] [74]:
- Leadership Commitment: Dedicating necessary human, financial, and technological resources.
- Accountability: Appointing a physician and pharmacist leader responsible for program outcomes.
- Action: Implementing interventions like prospective audit and feedback and preauthorization to optimize antibiotic use.
- Tracking and Reporting: Monitoring antibiotic prescribing and resistance patterns, and reporting this data to prescribers and leadership.
- Education: Informing prescribers and patients about antibiotic resistance and optimal prescribing.
Experimental Protocols for Resistance Research
Protocol 1: Evaluating Outer Membrane Permeabilizing Adjuvants
Objective: To assess the ability of a candidate adjuvant (e.g., SPR741) to sensitize a Gram-negative bacterium to a Gram-positive-specific antibiotic (e.g., rifampin) [3] [15].
Materials:
- Bacterial strain (e.g., E. coli ATCC 25922)
- Cation-adjusted Mueller-Hinton broth (CAMHB)
- Candidate adjuvant (e.g., SPR741 stock solution)
- Partner antibiotic (e.g., rifampin stock solution)
- 96-well sterile microtiter plates
- Automated broth microdilution system
Methodology:
- Broth Microdilution Checkerboard Assay:
- Prepare a 2D serial dilution of the adjuvant and the partner antibiotic in a 96-well plate, creating a checkerboard pattern with varying concentrations of each.
- Standardize the bacterial inoculum to ~5 × 10^5 CFU/mL and add to each well.
- Include growth control (bacteria only), sterility control (media only), and adjuvant toxicity control (adjuvant only at highest concentration).
- Incubate the plate at 35°C for 16-20 hours.
- Data Analysis:
- Determine the Minimum Inhibitory Concentration (MIC) of the antibiotic alone and in combination with each concentration of the adjuvant.
- The Fractional Inhibitory Concentration (FIC) index is calculated as:
FIC Index = (MIC of antibiotic in combination / MIC of antibiotic alone) + (MIC of adjuvant in combination / MIC of adjuvant alone) - Synergy is typically defined as an FIC Index of ≤0.5.
Protocol 2: Investigating Efflux Pump Activity
Objective: To determine the contribution of efflux pumps to a strain's resistance phenotype using an efflux pump inhibitor (EPI) like Phe-Arg-β-naphthylamide (PAβN).
Materials:
- Test and control bacterial strains
- Mueller-Hinton Agar (MHA) plates
- Antibiotic of interest (e.g., levofloxacin)
- EPI stock solution (e.g., PAβN)
- Blank antimicrobial disks
Methodology:
- Disk Diffusion Assay with EPI:
- Prepare a 0.5 McFarland standard suspension of the test bacterium and lawn it onto an MHA plate.
- Apply a disk impregnated with the antibiotic and a second disk impregnated with the antibiotic + EPI.
- Alternatively, place a blank disk containing the EPI adjacent (20-25 mm center-to-center) to the antibiotic disk to allow the inhibitors to diffuse together.
- Incubate at 35°C for 16-20 hours.
- Interpretation:
- Measure the zones of inhibition around both disks.
- An increase in the zone diameter by ≥5 mm around the disk containing the antibiotic + EPI (or in the area between the two disks) indicates significant efflux pump activity.
Research Reagent Solutions
Table 2: Essential Research Reagents for Studying Gram-Negative Resistance
| Reagent / Tool | Function / Application | Example Use |
|---|---|---|
| Polymyxin B Nonapeptide (PMBN) | Outer membrane permeabilizing adjuvant; benchmark compound [3] [15]. | Sensitizing E. coli to rifampin in checkerboard assays [15]. |
| SPR741 / SPR206 | Next-generation polymyxin-derived adjuvants with improved safety profiles; clinical-stage candidates [3]. | In vitro and in vivo models to potentiate partner antibiotics [3]. |
| Phe-Arg-β-naphthylamide (PAβN) | Broad-spectrum efflux pump inhibitor [8]. | Disk diffusion or MIC assays to confirm efflux-mediated resistance [8]. |
| Avibactam | Non-β-lactam β-lactamase inhibitor; targets Class A, C, and some D enzymes [15] [44]. | Used in combination with ceftazidime to restore susceptibility in KPC and AmpC producers [72] [44]. |
| Lytic Bacteriophages | Viruses that specifically infect and lyse bacterial hosts [55]. | Compassionate use therapy for biofilm-associated infections (e.g., P. aeruginosa in cystic fibrosis) [55]. |
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized medium for antimicrobial susceptibility testing (AST) [8]. | Performing broth microdilution for MIC determinations as per CLSI guidelines [72]. |
Signaling Pathways and Experimental Workflows
Diagram: Gram-Negative Bacterial Resistance Mechanisms
Diagram: Screening Workflow for Antibiotic Adjuvants
The fight against antimicrobial resistance (AMR), particularly in Gram-negative bacteria, represents a critical frontier in modern medicine. Gram-negative bacteria, such as Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa, are notably successful due to their intrinsic resistance mechanisms, which include low-permeability outer membranes and potent efflux pump systems [75]. This intrinsic resistance, coupled with their ability to rapidly acquire new resistance mechanisms, makes them a formidable threat in healthcare settings [75]. However, the scientific challenge of overcoming these biological barriers is compounded by a severe economic crisis in antibiotic development. The traditional drug development model, which relies on high sales volumes to recoup research and development (R&D) costs, fails for antibiotics because new agents must be used sparingly and held in reserve to slow the emergence of resistance [76] [77]. This has led to a market failure, where the development of new antibiotics is commercially unattractive, causing a collapse in the R&D pipeline despite overwhelming public health need [76] [77]. This article explores how "push" and "pull" incentives are designed to navigate this paradox, ensuring that our scientific arsenal can keep pace with evolving bacterial resistance within the specific context of Gram-negative pathogens.
FAQs: Understanding the Economic and Research Landscape
What are "push" and "pull" incentives and how do they differ?
In the context of antibiotic development, "push" and "pull" incentives are two complementary approaches to solve the market failure, but they target different stages of the drug development lifecycle.
Push Incentives: These aim to support the early, high-risk stages of innovation and R&D. Their primary goal is to lower the upfront costs and financial risks for developers, from basic science research through to clinical trials. This support is provided regardless of whether the product eventually reaches the market. Examples include direct research grants, public funding of preclinical work, and tax credits [78].
Pull Incentives: These aim to reward successful outcomes. They are designed to reduce the risk of insufficient future revenues by creating a viable market for new antibiotics that have proven to be scientifically viable and relevant. The goal is to ensure developers can achieve a financial return that justifies their investment, independent of high sales volumes. Mechanisms include market entry rewards, subscription-based models, and transferable exclusivity vouchers [78] [77].
The table below summarizes the key differences:
Table 1: Comparison of Push and Pull Incentives
| Feature | Push Incentives | Pull Incentives |
|---|---|---|
| Primary Goal | Lower R&D costs and risks | Ensure future revenue and market viability |
| Stage of Intervention | Early stages (research through clinical trials) | Late stages (upon regulatory approval or market entry) |
| Financial Flow | Downstream to developer (grants, subsidies) | Upstream to developer (rewards, guaranteed purchases) |
| Dependency | Not tied to market success | Contingent on successful development and approval |
| Example Mechanisms | Direct grants, tax credits, funding academic research | Market entry rewards, subscription models, milestone prizes |
Why is the standard commercial model failing for new antibiotics?
The commercial failure of new antibiotics is not due to a lack of efficacy, but rather a misalignment between public health needs and market forces. Key factors include:
- The Stewardship Imperative: To preserve the effectiveness of new antibiotics for as long as possible, they must be used judiciously and sparingly, immediately limiting their sales volume upon launch [76] [77]. This careful stewardship is at direct odds with the traditional pharmaceutical business model, which relies on high sales to generate revenue.
- Benchmarking Against Generics: New, innovative antibiotics are often priced in a market saturated with cheap, generic antibiotics [76]. Hospitals and payers are reluctant to pay a significant premium for a new drug, especially when older generics are still effective for many infections.
- Reimbursement Disincentives: In some healthcare systems, like the United States' Diagnosis-Related Group (DRG) system, hospitals receive a fixed payment for a patient's episode of care. If a hospital uses an expensive new antibiotic to treat a resistant infection, it may incur a financial loss, creating a powerful disincentive to stock new agents [76].
- High Cost and High Risk of Development: Developing a new antibiotic can take over a decade and cost more than $1 billion [77]. The scientific challenges are significant, with high failure rates. For investors, this high risk and low potential return make other therapeutic areas, like oncology or neurology, far more attractive [77].
What are the specific challenges in developing antibiotics against intrinsic resistance in Gram-negative bacteria?
Overcoming intrinsic resistance in Gram-negative bacteria presents unique and profound scientific hurdles that directly impact the economic calculus of development.
- The Permeability Barrier: The outer membrane of Gram-negative bacteria acts as a formidable physical barrier, greatly limiting the access of many antibiotic classes to their intracellular targets [75]. Designing molecules that can penetrate this membrane is a major challenge.
- Efflux Pump Systems: Gram-negative bacteria express powerful, broad-spectrum efflux pumps that can actively recognize and expel a wide range of antibiotic compounds from the cell, further reducing intracellular drug concentration [75]. A successful drug must either avoid recognition by these pumps or inhibit their function.
- Interplay of Mechanisms: Intrinsic resistance is rarely due to a single mechanism. The synergy between the low-permeability membrane and efflux pumps creates a powerful defensive network that is difficult to breach [75]. Furthermore, Gram-negative bacteria can rapidly acquire additional resistance genes, rendering a new drug ineffective in a short time [75]. This unpredictability adds to the commercial risk, as a developer cannot know how long their product will remain clinically useful.
How can my research team access push funding for early-stage projects?
Targeting push funding requires a strategic approach focused on demonstrating both scientific innovation and alignment with public health priorities.
- Identify Relevant Grant Programs: Actively monitor announcements from national and international bodies with a mandate for AMR, such as the National Institutes of Health (NIH), the Biomedical Advanced Research and Development Authority (BARDA), and similar entities in the European Union.
- Focus on Unmet Needs: Structure your research proposal to address pathogens or resistance mechanisms listed as "critical priority" by organizations like the WHO. Emphasize projects targeting Gram-negative bacteria with intrinsic and multidrug resistance.
- Leverage Public-Private Partnerships: Explore partnerships with non-profit organizations like the AMR Action Fund, which is designed to bridge the funding gap for later-stage clinical development by leveraging industry expertise and capital [77].
- Collaborate with Academia and Research Institutes: Push incentives often support foundational research. Building consortia that include academic institutions can provide access to government-funded grants and resources for basic and translational science.
Troubleshooting Guide: Integrating Economic and Scientific Strategies
Problem: High-throughput screening against Gram-negative pathogens is failing to identify novel compounds with good activity.
Potential Causes and Solutions:
- Cause: Compound Inability to Penetrate the Outer Membrane.
- Solution: Implement counterscreening assays early in your workflow. Use engineered strains of E. coli that hyper-express efflux pumps (e.g., AcrAB-TolC) or have defined porin deficiencies. A compound that loses activity in these strains likely has a penetration issue. Focus on chemical templates known to have better permeability in Gram-negative species.
- Cause: Efflux Pump-Mediated Exclusion.
- Solution: Employ efflux pump inhibitors (EPIs) in your assay systems. If the minimum inhibitory concentration (MIC) of your lead compound drops significantly in the presence of a sub-lethal concentration of an EPI like PaβN, efflux is a major contributor to the lack of activity. Consider co-developing an EPI or modifying the compound to evade efflux recognition.
- Cause: Target Inaccessibility.
- Solution: Utilize bacterial cytological profiling (BCP) or other mechanism-of-action screening tools to determine if your compound is even reaching its intended target. This can help you distinguish between compounds that fail due to lack of penetration versus those that reach the target but are ineffective.
Problem: Our lead compound is effective in vitro but lacks efficacy in animal models of Gram-negative infection.
Potential Causes and Solutions:
- Cause: Inadequate Pharmacokinetics/Pharmacodynamics (PK/PD).
- Solution: Conduct extensive PK/PD studies to understand the compound's absorption, distribution, metabolism, and excretion (ADME). The compound may not be reaching the infection site at a sufficient concentration for a long enough duration. Optimize the dosing regimen or chemically modify the compound to improve its half-life and tissue penetration.
- Cause: Host Factor Interference.
- Solution: Test the compound's stability and activity in the presence of serum proteins, as high protein binding can reduce free, active drug concentrations. The host immune environment or physiological conditions (e.g., pH, divalent cation concentration) at the infection site may also be inactivating the compound.
- Cause: Development of Adaptive Resistance.
- Solution: Inoculate animals from an infection cohort at various time points post-treatment to isolate bacteria. Perform MIC testing on these re-isolated bacteria to check for a transient, adaptive increase in resistance that may explain the efficacy drop in vivo [75].
Problem: Securing continued investment for our antibiotic program is challenging due to perceived commercial risk.
Potential Causes and Solutions:
- Cause: Lack of a Clear Value Proposition for Pull Incentives.
- Solution: Proactively generate data that demonstrates the value of your compound to payers and policymakers. This includes:
- Spectrum of Activity: Precisely define the range of Gram-negative pathogens your drug targets, emphasizing activity against WHO "critical priority" pathogens.
- Unique Mechanism: Highlight how your compound overcomes specific intrinsic resistance mechanisms (e.g., bypasses efflux, utilizes a novel porin).
- In-vivo Efficacy Data: Collect robust data in multiple, clinically relevant animal models.
- Resistance Propensity: Conduct in vitro serial passage experiments to model how quickly resistance might develop. A slower resistance development is a major value driver [76].
- Solution: Proactively generate data that demonstrates the value of your compound to payers and policymakers. This includes:
- Cause: Investor Preference for Other Therapeutic Areas.
- Solution: Actively engage with organizations and investors specializing in AMR. Structure your development plan to be "capital-efficient," using push funding for high-risk stages. Clearly articulate how your program is designed to be an attractive candidate for future pull incentives, such as a market entry reward.
Essential Experimental Protocols & Workflows
Protocol: Standardized Checkerboard Assay for Synergy Screening
Purpose: To identify synergistic interactions between a novel antibiotic and an efflux pump inhibitor (EPI) or another antibiotic against Gram-negative bacteria.
Materials:
- Bacterial Strain: Fresh overnight culture of the target Gram-negative bacterium (e.g., K. pneumoniae).
- Antimicrobials: Stock solutions of the novel antibiotic and the EPI (e.g., PaβN).
- Media: Cation-adjusted Mueller-Hinton Broth (CAMHB).
- Equipment: Sterile 96-well microtiter plates, multichannel pipettes, plate reader/incubator.
Methodology:
- Broth Preparation: Prepare a 2x concentration of CAMHB and distribute it into the 96-well plate.
- Compound Dilution:
- Serially dilute the novel antibiotic along the x-axis of the plate, typically achieving a 64x concentration range.
- Serially dilute the EPI along the y-axis of the plate over the same range.
- This creates a matrix where each well contains a unique combination of the two compounds at different concentrations.
- Inoculation: Dilute the bacterial overnight culture to ~1 x 10^6 CFU/mL in CAMHB and add an equal volume to each well, resulting in a final inoculum of ~5 x 10^5 CFU/mL and 1x compound concentrations.
- Incubation: Incubate the plate at 37°C for 18-24 hours.
- Analysis: Determine the Minimum Inhibitory Concentration (MIC) of each drug alone and in combination. The Fractional Inhibitory Concentration (FIC) index is calculated as:
FIC Index = (MIC of Drug A in combination / MIC of Drug A alone) + (MIC of Drug B in combination / MIC of Drug B alone)
- Synergy is typically defined as an FIC Index ≤ 0.5.
Protocol: Intrinsic Resistance Profiling Using Engineered Strains
Purpose: To deconvolute the contribution of specific intrinsic resistance mechanisms (efflux, permeability) in a Gram-negative bacterium.
Materials:
- Bacterial Panel: Isogenic strains differing in a single resistance mechanism. Example for E. coli:
- Wild-type strain (e.g., AG100).
- Efflux pump overexpressor (e.g., AG102, which expresses AcrAB from a marR mutation).
- Efflux pump knockout (e.g., AG100A, ΔacrAB).
- Porin-deficient mutant (e.g., a strain lacking OmpF and OmpC).
- Antimicrobials: Your lead compound and control antibiotics with known behavior (e.g., ciprofloxacin for efflux, meropenem for porins).
- Equipment: Materials for standard broth microdilution MIC testing.
Methodology:
- MIC Determination: Perform standard broth microdilution MIC assays according to CLSI/EUCAST guidelines against the entire panel of engineered strains.
- Data Interpretation:
- Efflux Impact: A ≥ 4-fold increase in the MIC of your compound in the overexpressor strain compared to the wild-type indicates it is a substrate for that efflux pump. A ≥ 4-fold decrease in the MIC in the knockout strain confirms this.
- Porin Dependence: A ≥ 4-fold increase in the MIC in the porin-deficient strain compared to the wild-type suggests your compound relies on that porin for entry.
- Visual Workflow: The following diagram illustrates the logical workflow for profiling intrinsic resistance mechanisms.
Diagram 1: Intrinsic resistance profiling workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for Gram-Negative AMR Research
| Reagent / Tool | Function / Application | Key Considerations |
|---|---|---|
| Engineered Bacterial Strains (e.g., efflux knockouts/overexpressors) | Deconvoluting specific resistance mechanisms (efflux, permeability). | Ensure strains are isogenic to the wild-type control to avoid confounding genetic differences. |
| Broad-Spectrum Efflux Pump Inhibitors (e.g., PaβN, CCCP) | Screening tool to identify if a compound is an efflux substrate and to potentiate activity. | Many are toxic for therapeutic use but are invaluable for in vitro mechanistic studies. |
| Cation-Adjusted Mueller-Hinton Broth (CAMHB) | Standardized medium for antibiotic susceptibility testing (AST). | Essential for reproducible MIC results as divalent cations can affect antibiotic activity, especially against P. aeruginosa. |
| Clinical and Laboratory Standards Institute (CLSI) Documents (e.g., M07, M100) | Provides standardized methodologies and breakpoints for AST. | Adherence to guidelines is critical for generating reliable, comparable data. EUCAST standards are an alternative. |
| Membrane Permeabilizers (e.g., Polymyxin B nonapeptide, EDTA) | Tools to chemically disrupt the outer membrane and study its barrier function. | Useful for understanding the contribution of the lipopolysaccharide layer to intrinsic resistance. |
| Real-Time PCR Assays | Quantifying expression levels of efflux pump and porin genes in response to antibiotic exposure. | Helps identify if resistance is mediated by upregulation of intrinsic mechanisms. |
| Bioluminescent or Fluorescent Reporter Strains | Visualizing compound penetration and accumulation in live bacteria in real-time. | Provides direct evidence of whether a compound is entering the cell and being effluxed. |
Bench to Bedside: Preclinical and Clinical Validation of Novel Therapeutics
The rising threat of antibiotic-resistant gram-negative bacteria represents a global health crisis, with the Centers for Disease Control and Prevention predicting that by 2050, more deaths will result from microorganisms than all cancers combined [79]. Effectively combating this threat requires robust, predictive models for evaluating new compounds against intrinsically resistant pathogens like Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae [79] [80]. Traditional models often fail to accurately replicate the in vivo bacterial environment, leading to poor correlation between in vitro and in vivo assays and limited therapeutic potential [79]. This technical support center provides targeted guidance for researchers navigating the challenges of efficacy testing against gram-negative bacteria with complex intrinsic resistance mechanisms, with content specifically framed within the context of overcoming intrinsic resistance in gram-negative bacterial research.
Frequently Asked Questions (FAQs)
Q1: Why do my in vitro antibiotic efficacy results often fail to predict in vivo outcomes?
Several factors contribute to this disconnect:
- Physiologically irrelevant growth conditions: Standard rich laboratory media (like CAMHB) lack key physiological components and do not mimic infection site environments [80]. For instance, M9 minimal medium with glucose better predicts lung infection outcomes for Klebsiella pneumoniae than standard media [80].
- Absence of fluid dynamics: Static culture conditions fail to account for fluid flow effects present in the body. Research shows that Pseudomonas aeruginosa displays different susceptibility patterns under flow conditions compared to static cultures [81].
- Biofilm formation limitations: Traditional planktonic cultures don't account for biofilm-mediated resistance, which can increase antibiotic tolerance by 10-1000-fold [79].
Q2: How can I better account for intrinsic resistance mechanisms in my efficacy models?
Gram-negative bacteria possess sophisticated intrinsic resistance mechanisms including:
- Impermeable outer membranes that limit antibiotic penetration [82] [83]
- Efflux pump systems (e.g., AcrAB-TolC in E. coli) that actively export antibiotics [82] [83]
- Enzymatic inactivation through chromosomally-encoded enzymes like β-lactamases [82] Incorporate assessment of these mechanisms by including efflux pump inhibitors, membrane permeabilizers, and isogenic mutant strains in your screening pipeline [83].
Q3: What advanced models can better predict clinical efficacy for intrinsically resistant pathogens?
Consider implementing these sophisticated approaches:
- Organ-on-a-chip and microfluidic systems that incorporate fluid flow and biomechanical cues [79] [81]
- Tissue mimetic media that better replicate infection site nutrition and biochemistry [80]
- Three-dimensional organotypic models that simulate healthy and diseased tissue states [84]
- Ex vivo systems using human tissue samples to maintain native architecture [79]
Q4: How can I "resistance-proof" new compounds against gram-negative pathogens?
Emerging strategies include:
- Exploiting resistance mechanisms against the pathogen, such as prodrugs activated by bacterial resistance enzymes [85]
- Targeting intrinsic resistance pathways like efflux pumps and cell envelope biogenesis to create hypersensitive strains [83]
- Combination therapies that simultaneously target multiple vulnerability points [80] [85]
- Evolutionary approaches that consider adaptive responses during compound development [83]
Troubleshooting Common Experimental Challenges
Problem: Inconsistent Antibiotic Combination Results Across Studies
Potential Causes and Solutions:
Table: Troubleshooting Antibiotic Combination Testing
| Issue | Root Cause | Solution |
|---|---|---|
| Variable synergy/antagonism reports | Different growth media affecting bacterial metabolism | Standardize using tissue-relevant media (e.g., M9Glu for lung infections) [80] |
| Poor translatability to animal models | Static culture conditions lacking physiological fluid flow | Implement microfluidic systems with controlled flow rates [81] |
| Species-specific variability | Differential expression of intrinsic resistance mechanisms | Include multiple Gram-negative species in screening panels [80] |
| Biofilm confounding results | Planktonic vs. biofilm susceptibility differences | Incorporate biofilm models in testing workflow [79] |
Problem: Overcoming Biofilm-Mediated Resistance in Testing
Biofilms dominate numerous chronic bacterial infections and are notoriously difficult to treat due to:
- Extracellular polymeric substance (EPS) matrix limiting antibiotic penetration [79]
- Metabolically heterogeneous subpopulations with varying susceptibility [79]
- Upregulation of efflux pumps and stress response pathways [79]
- Suppression of host immune responses [79]
Solutions:
- Implement biofilm-specific models such as flow cells, microtiter plate assays, or bioelectric effect systems [79]
- Include biofilm-disrupting agents in combination therapies [79]
- Extend treatment duration in efficacy studies to account for biofilm tolerance [79]
Problem: Addressing Evolutionary Resistance During Compound Development
Strategies to limit resistance emergence:
- High-throughput screening of knockouts (e.g., E. coli Keio collection) to identify intrinsic resistance genes [83]
- Experimental evolution under drug pressure to identify common resistance pathways [83]
- Resistance-proofing by targeting vulnerabilities that constrain evolutionary escape routes [83]
Table: Experimental Evolution Approaches for Resistance Assessment
| Method | Application | Outcome Measures |
|---|---|---|
| Serial passage at sub-MIC | Identify resistance development potential | MIC changes over generations [83] |
| Evolution in hypersensitive backgrounds | Test resistance-proofing strategies | Extinction frequency under drug pressure [83] |
| Whole-genome sequencing of evolved populations | Identify resistance mechanisms | Mutational signatures in resistant isolates [86] [83] |
| Efflux pump inhibition combined with experimental evolution | Assess adaptability to combination therapies | Frequency of dual resistance emergence [83] |
Standardized Experimental Protocols
Protocol 1: Microfluidic Antibiotic Efficacy Testing Under Flow Conditions
Background: Fluid flow significantly impacts antibiotic efficacy against Gram-negative pathogens. Pseudomonas aeruginosa shows increased susceptibility to antibiotics under flow conditions compared to static cultures [81].
Materials:
- Microfluidic device system with programmable flow controls
- Bacterial strains (e.g., P. aeruginosa, A. baumannii, K. pneumoniae)
- Antibiotic stock solutions
- Appropriate growth media (CAMHB, tissue-mimetic media)
- Syringe pumps and associated tubing
- Microscopy system for real-time monitoring
Procedure:
- Culture bacteria overnight and dilute to appropriate density (typically 10^5-10^6 CFU/mL)
- Load bacterial suspension into microfluidic device
- Program flow rates to mimic physiological conditions (e.g., 0.1-10 μL/min for capillary flow)
- Introduce antibiotic at clinically relevant concentrations
- Monitor bacterial viability in real-time using fluorescence markers or phase-contrast microscopy
- Compare results to static control conditions
- Quantify dose-response relationships under flow versus static conditions
Expected Outcomes: Research demonstrates that antibiotics previously classified as ineffective against resistant pathogens in static assays may show significant efficacy under flow conditions [81].
Protocol 2: Tissue-Mimetic Media for Predictive Susceptibility Testing
Background: Growth medium composition dramatically impacts antibiotic potency and drug-drug interactions. Tissue-mimetic media better predict in vivo outcomes than standard rich media [80].
Materials:
- Standard rich medium (CAMHB)
- Minimal medium (M9 salts with 0.5% glucose and 0.6 μM Fe(II)SO4, pH 7.0)
- Urine mimetic medium (for UTI models) [80]
- Test antibiotics
- Gram-negative ESKAPE pathogens
Procedure:
- Prepare the three media types: CAMHB, M9Glu, and urine mimetic medium
- Standardize bacterial inoculum across conditions
- Perform checkerboard assays or diagonal measurements of n-way drug interactions (DiaMOND) in each medium [80]
- Determine IC50 values and drug interaction indices (synergistic, additive, antagonistic)
- Compare results across media conditions
- Validate predictions in appropriate animal models
Expected Outcomes: Studies show that M9Glu medium better predicts in vivo efficacy in lung infection models compared to rich media, particularly for Klebsiella pneumoniae [80].
Signaling Pathways and Resistance Mechanisms
Intrinsic Resistance Pathways in Gram-Negative Bacteria
Experimental Workflow for Assessing New Compounds
Research Reagent Solutions
Table: Essential Research Reagents for Intrinsic Resistance Studies
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Tissue-Mimetic Media | M9Glu (M9 + 0.5% Glucose + 0.6 µM Fe(II)SO4), Urine Mimetic Medium [80] | Improved prediction of in vivo efficacy | M9Glu better predicts lung infection outcomes than rich media [80] |
| Efflux Pump Inhibitors | Chlorpromazine, Piperine, Verapamil [83] | Sensitization to multiple antibiotic classes | Rapid evolutionary adaptation to EPIs may occur [83] |
| Isogenic Mutant Strains | ΔacrB, ΔrfaG, ΔlpxM E. coli strains [83] | Identification of intrinsic resistance mechanisms | ΔacrB shows compromised ability to evolve resistance [83] |
| Microfluidic Systems | Programmable flow devices with bacterial culture chambers [81] | Physiological fluid flow conditions | Flow significantly increases antibiotic efficacy against resistant strains [81] |
| Biofilm Assessment Tools | Flow cells, Microtiter plate assays, EPS staining kits [79] | Biofilm-mediated resistance evaluation | Biofilms can tolerate 10-1000x higher antibiotic concentrations [79] |
| Whole Genome Sequencing | XGBoost-based ML models for resistance prediction [86] | Rapid resistance genotyping | Predicts resistance in A. baumannii, E. coli, K. pneumoniae [86] |
| Prodrug Compounds | Modified florfenicol exploiting Eis2 activation [85] | Resistance mechanism exploitation | Converts resistance proteins into drug activators [85] |
Frequently Asked Questions (FAQs)
Q1: My bacterial cultures are not evolving resistance in the morbidostat and are getting washed out. What could be wrong?
This is often due to an initial drug concentration that is too high, not allowing for initial adaptation. The drug concentration in the drug medium should be started from a level close to the minimum inhibitory concentration (MIC) of the ancestral strain. The algorithm should apply gentle, incremental selective pressure. Furthermore, ensure that the optical density threshold (OD_THR) is set appropriately (typically between 0.15 and 0.4) to prevent the culture from being washed out before mutations can emerge. If the OD is below this threshold, only drug-free media is added, allowing the population to recover [87] [88].
Q2: I am observing inconsistent growth rate measurements. How can I improve the reliability of my data? Inconsistent measurements can stem from biofilm formation on the inner walls of the vials or air bubbles in the culture due to insufficient mixing. Ensure the magnetic stirrer is functioning correctly and that the vial is being mixed continuously. The optical detection system should take measurements at frequent intervals (e.g., every 1-2 seconds) and average them over a short period (e.g., 1 minute) to smooth out noise. Also, verify that the LED light source and photo-detector are correctly aligned and clean [87].
Q3: The calculated drug concentration in the vial is becoming inaccurate over time. How can this be fixed?
The morbidostat calculates the drug concentration based on the dilution history. Inaccuracies can accumulate from small, impractical dosing volumes or from manually removing samples without updating the internal volume state. To address this, set a minimum_dosing_volume_ml parameter (e.g., 0.1 mL) in the control software; any calculated volume below this threshold is set to zero. Additionally, if you sample from the vial, ensure your software can account for the removed volume to maintain accurate concentration and volume tracking [89].
Q4: What does it mean if my evolved populations show different mutations but similar resistance levels?
This indicates convergent evolution, where different genetic solutions lead to the same resistant phenotype. This is a common and powerful finding in morbidostat experiments. For example, in the case of ciprofloxacin, different mutations in the target genes gyrA or gyrB can independently confer similar levels of resistance. This highlights the robustness of the resistance landscape and identifies key resistance "hotspots" [90] [91].
Troubleshooting Guides
Problem: Failure to Maintain Constant Growth Inhibition
Symptoms
- Bacterial culture optical density (OD) consistently exceeds or drops below the target threshold.
- The drug concentration in the culture vials does not increase steadily over time.
Solutions
- Check Algorithm Parameters: Verify the settings for the dilution rate, target OD, and growth rate. The dilution rate should be set significantly lower (e.g., half) than the maximal growth rate of the bacteria to force the system to adjust drug concentration [87].
- Calibrate Optical Density Sensors: Regularly calibrate the LED and photo-detector system against a known standard to ensure accurate OD measurements.
- Verify Drug Stock Concentration: Confirm the concentration and stability of the antibiotic in the drug reservoir. Prepare fresh stock solutions if necessary.
- Review PID Settings: If using a PID controller, the parameters may be too aggressive. Tune the PID values (Kp, Ki, Kd) to prevent oscillations in OD and ensure stable control. A Ziegler-Nichols approach can be used for tuning [89].
Problem: Contamination or Biofilm Formation
Symptoms
- Unusual changes in growth patterns or morphology under the microscope.
- Visible film or clumps on the walls of the culture vial.
Solutions
- Sterilization Protocol: Ensure all glass vials, tubing, and connectors are properly sterilized by autoclaving before use. Silicone and PTFE tubing can be further sterilized with Virkon and ethanol, followed by rinsing with sterile water [88].
- System Design: Use a Teflon insert with secure sealing for all tubing inlets and outlets to maintain a closed system.
- Regular Sampling and Inspection: Periodically sample the culture and inspect for contamination using plating and microscopy.
Experimental Protocols & Workflows
Standardized Morbidostat Experimental Evolution Protocol
The following protocol provides a detailed methodology for running a morbidostat experiment to study the evolution of antibiotic resistance in Gram-negative bacteria.
1. Equipment and Reagent Setup
- Morbidostat Device: Assemble the system, including culture vials, peristaltic pumps, optical density sensors, and a control unit (e.g., Arduino/Raspberry Pi) [87] [91].
- Culture Vials: Use flat-bottom glass vials with a working volume of 12-20 mL. Equip each vial with a magnetic stir bar for continuous mixing [87].
- Tubing and Connectors: Use autoclavable PEEK or silicone tubing for all fluid paths. Employ Luer thread-style connectors for secure, leak-proof connections [87].
- Media and Drugs: Prepare sterile growth media (e.g., LB or Mueller-Hinton Broth). Prepare a concentrated stock of the antibiotic (e.g., 10x the desired starting concentration) in sterile media or an appropriate solvent [92] [88].
2. Initialization and Inoculation
- Sterilization: Autoclave all glass vials, stir bars, and tubing assemblies before the experiment [88].
- Inoculation: Thaw frozen bacterial stock (e.g., Acinetobacter baumannii, Escherichia coli, Pseudomonas aeruginosa) and transfer it to the morbidostat vial containing fresh, sterile medium to achieve an initial 60-fold dilution [88].
- System Start-Up: Place the inoculated vials into the holder, start the magnetic stirrer, and begin the control software. Initialize the algorithm with the required parameters from the table below.
3. Morbidostat Operation and Monitoring
- The control software continuously monitors the OD and, at fixed intervals (∆t, typically 12 minutes), decides to add a fixed volume (∆V, typically 1 mL) of either fresh media or drug media [87] [88].
- The algorithm for this decision is based on the following logic, which can be visualized in the workflow diagram below:
4. Sampling and Endpoint Analysis
- Population Sampling: Regularly collect population samples (e.g., daily) from each vial for glycerol stock storage at -80°C and subsequent genomic analysis [88] [91].
- Clonal Analysis: At the end of the experiment, isolate individual clones from the evolved populations. Determine their MIC to the antibiotic and subject them to whole-genome sequencing to identify resistance-conferring mutations [90] [91].
- Data Processing: Use bioinformatics pipelines to process deep sequencing data from population time-series samples, identifying single-nucleotide variants (SNVs), insertions/deletions (indels), and genomic rearrangements [91].
Key Experimental Parameters for Morbidostat Operation
The table below summarizes the critical parameters for setting up a morbidostat experiment, with typical values derived from the literature.
| Parameter | Symbol | Typical Value | Function and Impact |
|---|---|---|---|
| Culture Volume | V |
12 - 20 mL | Total volume of the bacterial culture in the vial. |
| Dilution Volume | ∆V |
1 mL | Fixed volume of media added in each dilution event. |
| Dilution Interval | ∆t |
12 minutes | Fixed time interval between dilution events. |
| Dilution Rate | r_dilution |
~0.4 hr⁻¹ | ∆V/(V * ∆t). Sets the target growth rate for the inhibited culture. |
| OD Threshold | OD_THR |
0.15 - 0.4 | Threshold OD above which drug media may be added. Prevents washout. |
| Initial Drug Conc. | - | 1x - 10x MIC | The concentration of antibiotic in the initial drug medium. |
Research Reagent Solutions
The following table lists essential materials and reagents commonly used in resistomics studies involving morbidostats and Gram-negative bacteria.
| Reagent/Material | Function in Experiment | Specific Examples & Notes |
|---|---|---|
| Gram-Negative Bacterial Strains | Model organisms for experimental evolution. | ESKAPE pathogens: Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli [90] [8] [92]. |
| Antibiotics | Selective pressure driving evolution. | Ciprofloxacin, Triclosan, TGV-49. Used in a range of concentrations to study resistance pathways [90] [92] [91]. |
| Growth Media | Supports bacterial growth in continuous culture. | Lysogeny Broth (LB), Mueller-Hinton Broth (MHB). Must be compatible with the organism and the antibiotic [92] [91]. |
| Morbidostat Culture Vials | Container for growing bacterial cultures. | Flat-bottom glass vials with open-top screw caps and Teflon inserts for tubing [87]. |
| Autoclavable Tubing | Transport of media and drugs to the culture vials. | PEEK (Polyether ether ketone) or silicone tubing. Must withstand autoclaving and resist chemical degradation [87]. |
| Novel Antimicrobial Agents | Testing compounds with low resistance potential. | TGV-49: A novel antimicrobial that disrupts the microbial membrane, showing minimal resistance development in a morbidostat [92]. |
| Adjuvants / Permeabilizers | Compounds that overcome intrinsic resistance by disrupting the outer membrane. | Polymyxin B nonapeptide (PMBN), SPR741. Used to sensitize bacteria to other antibiotics [10] [3]. |
The intrinsic resistance of Gram-negative bacteria to many antibiotic classes represents a significant hurdle in modern antimicrobial therapy. This resistance is predominantly due to the formidable barrier provided by the complex cell envelope, particularly the impermeable outer membrane and the presence of efflux pumps [3] [15]. Infections caused by multidrug-resistant Gram-negative pathogens such as Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae are associated with increased mortality rates and pose one of the most urgent threats to global healthcare [93]. This technical support center provides troubleshooting guidance and methodological support for researchers developing strategies to overcome these resistance mechanisms, with a particular focus on comparative analyses between novel therapeutic agents and conventional therapies.
Troubleshooting Guides & FAQs
Q1: My high-throughput screening assay for identifying potential antibiotic adjuvants is showing high variability in optical density (OD) readings. How can I improve assay robustness?
A: High variability often stems from inconsistent incubation conditions or bacterial preparation.
- Repeat the experiment: First, rule out simple procedural errors like incorrect pipetting volumes or unintended extra wash steps [28].
- Verify assay robustness using the Z' factor: Calculate the Z' factor, a standard parameter for assessing high-throughput screening assay quality. A Z' value of at least 0.5 is considered robust, while a value greater than 0.7 is excellent [94].
- Formula: Z’=1−[(3σneg+3σpos) / |μneg−μpos| ] where σ is the standard deviation and μ is the mean of the positive (pos, growth inhibited) and negative (neg, normal growth) controls [94].
- Procedure: Use half of a 384-well plate for each control condition to ensure adequate sample size. Use a positive control compound like erythromycin for S. aureus to inhibit growth [94].
- Check reagents and equipment: Ensure bacterial culture media like Tryptic Soy Broth (TSB) is fresh and properly stored. Verify that the plate reader is calibrated and that 384-well clear-bottom plates are handled consistently [94] [95].
Q2: I am testing a new polymyxin derivative as a permeabilizing adjuvant, but my positive control (Polymyxin B Nonapeptide, PMBN) is not producing the expected synergy with rifampin against a clinical isolate of E. coli. What could be wrong?
A: Failure of the positive control suggests a fundamental issue with the experimental conditions.
- Confirm bacterial strain integrity: The strain may have acquired resistance. Check its genotype and phenotype, particularly regarding lipid A modification pathways (e.g., pmrCAB or arn operon) that can lower the negative charge of LPS and reduce polymyxin binding [3].
- Check adjuvant and antibiotic stocks: Ensure the PMBN and antibiotic stocks are prepared correctly, have not expired, and have been stored at the appropriate temperature. Contaminated or degraded reagents are a common cause of failure [95].
- Re-optimize concentrations: The concentrations of both the adjuvant and the partner antibiotic are critical. Set up a checkerboard assay with a range of concentrations for both compounds to confirm the expected synergistic interaction and re-establish the minimum inhibitory concentration (MIC) for your system [15].
Q3: When preparing biological media bases for susceptibility testing, I am observing precipitation or crystallization. How can I resolve this?
A: Precipitation is typically related to improper dissolution or a reaction between components.
- Follow preparation instructions meticulously: Adhere strictly to the manufacturer's protocols regarding water quality, temperature, and mixing order [95].
- Ensure thorough mixing: Use a sterile magnetic stirrer or vortex mixer to achieve complete homogeneity and dissolution of the media base [95].
- Discard and prepare a fresh batch: If precipitation or crystallization has occurred, it is recommended to discard the media and prepare a new batch to ensure accurate and reliable results. Using compromised media can lead to false negatives or inconsistent data [95].
Quantitative Data Comparison
Table 1: Comparative Efficacy of Selected Antibiotic Agents and Adjuvants Against Gram-Negative Bacteria
| Agent / Strategy | Mechanism of Action | Target Bacteria | Key Efficacy Metric | Performance vs. Conventional Therapy | Key Limitations |
|---|---|---|---|---|---|
| Octapeptin C4 [3] | Permeabilizes outer membrane | MDR Gram-negative (including polymyxin-resistant strains) | ~4-fold decrease in activity after resistance studies (vs. 1000-fold for polymyxin B) | Superior resistance profile compared to conventional polymyxins [3] | Still in preclinical investigation [3] |
| SPR741 / SPR206 [3] | Permeabilizes outer membrane (polymyxin derivative) | MDR Gram-negative | Synergy with rifampin/clarithromycin in murine models; reduced cytotoxicity | Shows promise as a safer, synergistic adjuvant compared to nephrotoxic polymyxins [3] | Phase I trials completed; clinical efficacy pending [3] |
| Dephostatin [3] | Disrupts PmrAB two-component signaling system | Polymyxin-resistant Gram-negative | Prevents lipid A modification, re-sensitizing bacteria to polymyxins | Novel target; overcomes acquired colistin resistance [3] | Mechanism and efficacy in complex models requires further validation [3] |
| Combination Therapy (Polymyxin + other antibiotic) [15] | Synergy via membrane permeabilization | MDR A. baumannii, P. aeruginosa | Variable synergy in clinical studies | A mainstay for infections with no other options [15] | Nephrotoxicity remains a concern; efficacy not always predictable [15] |
Table 2: High-Throughput Screening Parameters for Identifying Pathway-Specific Inhibitors
| Parameter | Specification | Purpose & Rationale |
|---|---|---|
| Paired Strains [94] | Wildtype + Pathway-null mutant | Identifies compounds that selectively inhibit conditionally essential enzymes in the target pathway. |
| Readout [94] | Optical Density (OD600) | Measures bacterial growth inhibition in a high-throughput manner. |
| Assay Robustness [94] | Z' factor ≥ 0.5 | Ensures the assay is reliable enough for screening; Z' > 0.7 is excellent. |
| Recommended Replication [94] | Duplicate runs of entire screen | Reduces false positive rates by as much as 50%. |
| Positive Control [94] | Broad-spectrum antibiotic (e.g., Erythromycin) | Validates growth inhibition conditions in each assay plate. |
| Biosafety [94] | BSL1 strains recommended; BSL2 requires containment | Ensures safety during automated screening procedures. |
Experimental Protocols
Basic Protocol: High-Throughput Screening for Growth-Inhibitory Compounds Using Paired Bacterial Strains
This protocol is designed to discover small molecules that specifically target conditionally essential enzymes within virulence factor biosynthetic pathways (e.g., teichoic acid biosynthesis) by screening against paired bacterial strains: a wildtype and a pathway-null mutant [94].
Materials:
- Bacterial Strains: Wildtype and isogenic mutant with the first enzyme in the targeted non-essential pathway inactivated [94].
- Growth Medium: Tryptic Soy Broth (TSB) or other appropriate medium [94].
- Equipment: 384-well clear-bottom plates, low-evaporation lids, pin transfer robot, multi-channel pipette, incubator, plate reader capable of OD600 measurements [94].
- Reagents: Chemical library, positive control antibiotic (e.g., erythromycin at 10 mg/mL in ethanol), DMSO (solvent control) [94].
Method:
- Inoculate and Grow Cultures: Inoculate 2 mL of TSB media with a single colony of each bacterial strain (wildtype and mutant) and grow to saturation by shaking at 30°C overnight [94].
- Dilute Cultures: Dilute the saturated cultures 1:1000 in fresh TSB to prepare the working inoculum [94].
- Pin Transfer Compounds: Using a pin transfer robot, transfer library compounds (nL volumes) from stock plates into the 384-well assay plates. Include controls: positive control (antibiotic) and negative control (DMSO) on each plate [94].
- Inoculate Assay Plates: Dispense 50 μL of the diluted bacterial inoculum into each well of the assay plates using a multi-channel pipette or liquid dispenser. For paired screening, each compound is tested against both the wildtype and mutant strains in separate plates [94].
- Incubate and Measure: Seal plates with low-evaporation lids. Incubate static at 30°C for 18-24 hours. Measure the optical density at 600 nm (OD600) as a proxy for bacterial growth [94].
- Data Analysis: Identify screening "hits" as compounds that inhibit growth of the wildtype strain but do not inhibit the growth of the pathway-null mutant. These compounds are likely targeting the conditionally essential enzymes in the pathway of interest [94].
Troubleshooting: If contamination is suspected, add selective antibiotics to the media during both pin transfer and bacterial inoculation steps [94].
Visualization of Concepts and Workflows
Diagram 1: Gram-Negative Cell Envelope & Resistance
Diagram 2: Paired Strain Screening Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Investigating Intrinsic Resistance
| Reagent / Material | Function in Research | Key Considerations |
|---|---|---|
| Dehydrated Culture Media (e.g., TSB) [94] [95] | General bacterial cultivation and growth in susceptibility assays. | Store in a cool, dry place. Discard if clumping occurs due to moisture. Ensure complete dissolution during preparation [95]. |
| 384-Well Clear-Bottom Plates [94] | Vessel for high-throughput growth inhibition assays compatible with OD readings. | Use with low-evaporation lids to prevent volume loss during incubation. |
| Selective Antibiotics [94] | Maintain plasmid-borne markers and prevent bacterial contamination during screening. | Add to media during both pin transfer and bacterial inoculation steps. |
| Pathway-Specific Paired Strains [94] | Wildtype and isogenic mutant to identify conditionally essential enzyme inhibitors. | Confirm conditional essentiality of the target pathway before screening. |
| Positive Control Antibiotics (e.g., Erythromycin) [94] | Validate growth inhibition conditions and assay performance on each plate. | Prepare fresh stocks and store appropriately to maintain stability. |
| Outer Membrane Permeabilizers (e.g., PMBN) [3] [15] | Positive control adjuvant for synergy studies with conventional antibiotics. | Check for loss of activity due to improper storage or age; known to re-sensitize bacteria to hydrophobic antibiotics [15]. |
The World Health Organization's (WHO) 2025 analysis of the antibacterial development pipeline reveals a fragile and contracting landscape. With only 90 antibacterial agents in clinical development globally (down from 97 in 2023), the pipeline remains insufficient to address the escalating threat of antimicrobial resistance, particularly for Gram-negative pathogens [96]. Of these candidates, only 50 are traditional antibiotics while 40 employ non-traditional approaches [96]. Most concerning is the limited innovation targeting critical priority pathogens, with only 5 of the 15 agents classified as "innovative" directed against WHO critical priority pathogens [96]. This review provides a critical assessment of these clinical-stage candidates within the specific context of overcoming intrinsic resistance in Gram-negative bacteria, offering technical guidance for researchers navigating this challenging field.
FAQ: Understanding the WHO Clinical Pipeline Data
What does the WHO clinical pipeline analysis encompass? The WHO's "Analysis of antibacterial agents in clinical and preclinical development: overview and analysis 2025" provides a comprehensive evaluation of the global antibacterial pipeline. This seventh clinical review examines both traditional (direct-acting small molecules) and non-traditional antibacterial candidates in development worldwide. The analysis specifically evaluates how effectively the current pipeline addresses infections caused by priority pathogens, as defined by the updated 2024 WHO bacterial priority pathogens list [97].
How does the pipeline address intrinsic resistance in Gram-negative bacteria? Intrinsic resistance in Gram-negative bacteria, primarily conferred by their unique cell envelope structure, represents a fundamental challenge in antibiotic development. The Gram-negative outer membrane, with its asymmetric bilayer containing lipopolysaccharide (LPS), acts as a formidable permeability barrier that prevents many antibiotics from reaching their intracellular targets [3] [15]. The current clinical pipeline contains candidates attempting to overcome this barrier through various strategies, including outer membrane disruption, efflux pump inhibition, and novel compound classes designed to bypass traditional penetration issues [97] [15].
What are the most significant gaps in the current clinical pipeline? Critical gaps identified in the 2025 WHO analysis include insufficient oral therapies for outpatient use, limited pediatric formulations, and concerningly few truly innovative agents targeting the highest-priority Gram-negative pathogens (Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae) [96]. The pipeline's fragility is further highlighted by the dominance of small, micro-sized entities driving development, creating significant volatility in the R&D ecosystem [96].
How is "innovation" defined in the WHO assessment? For traditional agents, the WHO evaluates innovation based on specific criteria: absence of known cross-resistance, new targets, novel modes of action, and/or new drug classes [97]. These criteria are particularly relevant for overcoming intrinsic resistance, as compounds meeting them are more likely to bypass the conventional permeability barriers that limit existing antibiotic classes [97] [15].
Table 1: WHO 2025 Clinical Pipeline Overview
| Pipeline Category | Number of Agents | Key Characteristics | Notable Gaps |
|---|---|---|---|
| Total Clinical Agents | 90 | Down from 97 in 2023; includes both traditional and non-traditional approaches | Fragile pipeline with limited commercial investment |
| Traditional Antibiotics | 50 | Direct-acting small molecules; many are derivatives of existing classes | Few address intrinsic resistance mechanisms in Gram-negative bacteria |
| Non-Traditional Approaches | 40 | Includes bacteriophages, antibodies, immunomodulators, and other novel modalities | Regulatory pathways for many approaches remain undefined |
| Innovative Agents | 15 | Meet WHO criteria for innovation (new targets, mechanisms, etc.) | Only 5 target WHO critical priority pathogens |
| Agents Targeting Critical Priority Pathogens | 5 | Focus on Acinetobacter, Pseudomonas, and Enterobacteriaceae | Insufficient to address current and anticipated resistance trends |
Table 2: Analysis of Clinical Candidates by Pathogen Priority
| WHO Pathogen Priority Level | Pathogen Examples | Number of Clinical Candidates | Development Challenges |
|---|---|---|---|
| Critical | Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacteriaceae | 5 innovative agents | Overcoming multidrug resistance including carbapenem resistance |
| High | Helicobacter pylori, Campylobacter spp., Salmonella spp. | Limited data in public domain | Need for narrow-spectrum agents with novel mechanisms |
| Medium | Streptococcus pneumoniae, Haemophilus influenzae, Shigella spp. | Multiple candidates in development | Balancing spectrum of activity with resistance concerns |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Intrinsic Resistance
| Reagent / Tool | Function / Application | Research Utility |
|---|---|---|
| Polymyxin B nonapeptide (PMBN) | Outer membrane permeabilizer without bactericidal activity | Useful for studying membrane permeability and potentiating other antibiotics [3] [15] |
| SPR741/NAB741 | Polymyxin B analogue with sensitizing activity | Completed Phase I trials; research tool for synergy studies with other antibiotics [3] |
| Octapeptin C4 | Cyclic peptide that permeabilizes outer membrane | Shows activity against polymyxin-resistant strains; valuable for studying alternative permeabilization strategies [3] |
| Dephostatin | Small molecule inhibitor of two-component systems (e.g., PmrAB) | Research tool for studying regulatory pathways involved in polymyxin resistance [3] |
| Porin Expression Assays | Methods to analyze outer membrane protein composition | Essential for studying antibiotic penetration pathways in Gram-negative bacteria |
| Efflux Pump Inhibitors | Compounds that block multidrug efflux systems | Research tools for determining efflux contribution to intrinsic resistance |
Experimental Protocols: Key Methodologies
Protocol 1: Assessing Outer Membrane Permeabilization as an Adjuvant Strategy
Background: The outer membrane of Gram-negative bacteria represents a significant barrier to antibiotic penetration. This protocol describes methodology for evaluating candidate compounds that disrupt outer membrane integrity, potentially restoring activity of otherwise ineffective antibiotics [3] [15].
Materials:
- Bacterial strains: Reference strains and clinical isolates of target Gram-negative pathogens
- Test antibiotics: Erythromycin, rifampin, novobiocin, clindamycin, vancomycin
- Candidate permeabilizers: PMBN, SPR741, octapeptin derivatives, or novel compounds
- Culture media: Mueller-Hinton broth and agar
- Equipment: Microdilution trays, incubator, spectrophotometer
Procedure:
- Prepare standardized bacterial inocula (approximately 5 × 10^5 CFU/mL) in suitable medium
- Conduct checkerboard broth microdilution assays with serial dilutions of candidate permeabilizer and partner antibiotic
- Include controls: bacteria alone, permeabilizer alone, antibiotic alone
- Incubate at 35°C for 16-20 hours
- Determine minimum inhibitory concentrations (MICs) visually or spectrophotometrically
- Calculate fractional inhibitory concentration (FIC) indices to quantify synergy
- Confirm membrane damage using 1-N-phenylnaphthylamine (NPN) uptake assay
- Validate findings with time-kill studies against relevant Gram-negative pathogens
Troubleshooting:
- Lack of observed synergy may indicate insufficient permeabilization or alternative resistance mechanisms
- Cytotoxicity screening is essential for any promising permeabilizer candidates
- Consider pathogen-specific differences in outer membrane composition that may affect results
Protocol 2: Evaluation of Efflux Pump Inhibition
Background: Efflux pumps contribute significantly to intrinsic resistance in Gram-negative bacteria by reducing intracellular antibiotic concentrations. This protocol details assessment of potential efflux pump inhibitors [15] [8].
Materials:
- Bacterial strains with characterized efflux systems
- Substrates for specific efflux pumps (e.g., ethidium bromide, Hoechst 33342)
- Candidate efflux pump inhibitors (e.g., PAβN, CCCP, novel compounds)
- Fluorometric detection equipment
- Culture media and antibiotics affected by efflux
Procedure:
- Grow bacterial cultures to mid-logarithmic phase in appropriate medium
- Assess accumulation of fluorescent substrate in presence and absence of inhibitor
- Measure efflux activity by pre-loading cells with substrate, adding inhibitor, and monitoring fluorescence decrease over time
- Conduct antibiotic susceptibility testing with and without subinhibitory concentrations of efflux pump inhibitor
- Evaluate impact on norfloxacin, chloramphenicol, and β-lactam MICs
- Use real-time PCR to assess potential impact on efflux pump gene expression
- Determine frequency of resistance emergence to inhibitor-antibiotic combinations
Troubleshooting Guide: Common Experimental Challenges
Problem: Lack of Synergy Between Candidate Adjuvant and Partner Antibiotic
Potential Causes and Solutions:
- Insufficient permeabilization: Titrate adjuvant concentration, ensuring it remains below standalone MIC while achieving effective membrane disruption. Confirm membrane activity with NPN uptake assay [3] [15].
- Alternative resistance mechanisms: The bacterium may possess additional resistance mechanisms beyond membrane permeability (e.g., drug inactivation, target modification). Conduct additional mechanistic studies to identify the primary resistance barrier [8].
- Inappropriate antibiotic selection: Verify that the antibiotic's cellular target is present and susceptible in the test organism. Some "Gram-positive only" antibiotics lack targets in Gram-negative bacteria entirely [15].
- Methodological issues: Ensure proper checkerboard assay technique with appropriate controls. Confirm bacterial inoculum purity and viability throughout the experiment [28].
Problem: High Cytotoxicity of Membrane-Active Compounds
Potential Causes and Solutions:
- Non-selective membrane activity: The compound may disrupt eukaryotic membranes similarly to bacterial membranes. Consider structural modification to increase selectivity for bacterial membranes [3] [15].
- Excessive positive charge: Many membrane-active compounds are cationic. Reducing net positive charge may decrease mammalian cell toxicity while retaining anti-Gram-negative activity [15].
- Suboptimal physicochemical properties: Modify logP, molecular weight, or other properties to improve selectivity index. SPR741 represents a successful example of such optimization [3].
Problem: Variable Activity Across Gram-Negative Species
Potential Causes and Solutions:
- Species-specific outer membrane differences: LPS structure, porin composition, and efflux systems vary significantly between Gram-negative species. Characterize the specific resistance mechanisms in each test organism [3] [8].
- Differential expression of resistance elements: Regulatory pathways controlling membrane permeability may vary. Assess gene expression of relevant regulators (e.g., PmrAB, PhoPQ) across species [3].
- Method standardization: Ensure consistent growth conditions, as membrane composition can vary with growth phase and medium composition [28].
Visualization: Experimental Workflows and Resistance Mechanisms
Diagram Title: Antibiotic Adjuvant Screening Workflow
Diagram Title: Gram-Negative Antibiotic Resistance Pathways
The WHO's 2025 clinical pipeline analysis reveals continued concerning trends in antibacterial development, with particular vulnerabilities in addressing intrinsic resistance mechanisms of Gram-negative pathogens. The decline in total clinical-stage agents (from 97 to 90) alongside the minimal number of innovative candidates targeting critical priority pathogens underscores the precarious state of antibacterial R&D [96]. Researchers focused on overcoming intrinsic resistance must prioritize strategies that directly address the fundamental permeability barriers of the Gram-negative cell envelope, including outer membrane disruption, efflux inhibition, and compound design informed by accumulation rules [15]. The experimental frameworks and troubleshooting guidance provided herein offer practical methodologies for advancing these critical efforts, with the ultimate goal of expanding therapeutic options against multidrug-resistant Gram-negative infections.
Conclusion
Overcoming intrinsic resistance in Gram-negative bacteria demands a multi-pronged, innovative approach that moves beyond traditional antibiotic discovery. The integration of foundational knowledge of bacterial cell envelope biology with advanced methodologies—such as adjuvant therapy, nanotechnology-enabled delivery, and novel membrane-disrupting agents—represents a paradigm shift in our antimicrobial arsenal. While significant challenges in economic viability and rapid resistance emergence remain, the convergence of these strategies offers a promising path forward. Future success will hinge on sustained global commitment, enhanced push-pull incentives for antibiotic development, and the continued application of advanced tools like AI and resistomics to design smarter, more durable therapeutics that can outmaneuver bacterial evolution and secure a future against drug-resistant infections.
References
- 1. The Beauty of Asymmetric Membranes - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC7381204/]
- 2. Biochemistry, Lipopolysaccharide - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK554414/]
- 3. Overcoming Intrinsic and Acquired Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7558195/]
- 4. Porins and specific channels of bacterial outer membranes [https://pubmed.ncbi.nlm.nih.gov/1373213/]
- 5. Porin (protein) [https://en.wikipedia.org/wiki/Porin_(protein)]
- 6. Structure and function of the porin channel [https://www.sciencedirect.com/science/article/pii/S0085253815591461]
- 7. Lipopolysaccharide [https://en.wikipedia.org/wiki/Lipopolysaccharide]
- 8. Evaluation of Antibiotic Resistance Mechanisms in Gram ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10668847/]
- 9. Mechanism of antibacterial resistance, strategies and next- ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1444781/full]
- 10. Overcoming Intrinsic Resistance in Gram-negative Bacteria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9885958/]
- 11. Role of bacterial efflux pumps in antibiotic resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10268834/]
- 12. Efflux pump-mediated resistance to new beta lactam ... [https://www.nature.com/articles/s43856-024-00591-y]
- 13. An overview of the antimicrobial resistance mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6604941/]
- 14. Intrinsic antibiotic resistance: Mechanisms, origins ... [https://www.sciencedirect.com/science/article/abs/pii/S1438422113000246]
- 15. Overcoming intrinsic resistance in gram-negative bacteria ... [https://www.sciencedirect.com/science/article/abs/pii/S0960894X22005893]
- 16. Emerging Strategies to Combat ESKAPE Pathogens in the Era ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6452778/]
- 17. Mechanisms of Antimicrobial Resistance in ESKAPE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4871955/]
- 18. New approaches for combating polyresistant ESKAPE pathogens [https://iimmun.ru/iimm/article/view/17784]
- 19. Antimicrobial resistance patterns of WHO priority ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11846185/]
- 20. Resistance mechanisms and therapeutic strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576158/]
- 21. Overcoming Intrinsic and Acquired Resistance ... [https://www.mdpi.com/2079-6382/9/9/623]
- 22. ESKAPE pathogens rapidly develop resistance against ... [https://www.nature.com/articles/s41564-024-01891-8]
- 23. The Global Burden of Multidrug-Resistant Bacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12101290/]
- 24. The global economic burden of antibiotic-resistant infections ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12182023/]
- 25. Economic costing methodologies for drug-resistant bacterial ... [https://healtheconomicsreview.biomedcentral.com/articles/10.1186/s13561-025-00644-5]
- 26. Current economic and regulatory challenges in developing ... [https://www.nature.com/articles/s44259-025-00123-1]
- 27. The Chemistry Center for Combating Antibiotic Resistant ... [https://www.rti.org/impact/novel-solutions-chemistry-center-combating-antibiotic-resistant-bacteria-cc4carb]
- 28. Troubleshooting Protocols - Biological Sciences: Summer ... [https://guides.library.cmu.edu/biologicalsciencesSRI/troubleshooting]
- 29. Gram-Negative Bacteria - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK538213/]
- 30. Antibiotic adjuvants against multidrug-resistant Gram- ... [https://www.sciencedirect.com/science/article/pii/S094450132400243X]
- 31. Antimicrobial resistance [https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance]
- 32. The Global Challenge of Antimicrobial Resistance [https://pmc.ncbi.nlm.nih.gov/articles/PMC12284435/]
- 33. Antibiotic potentiators as a promising strategy for ... [https://www.nature.com/articles/s44259-025-00112-4]
- 34. Strategies to Overcome Antimicrobial Resistance (AMR) ... [https://www.mdpi.com/1422-0067/20/23/5844]
- 35. Tackling the outer membrane: facilitating compound entry ... [https://www.nature.com/articles/s44259-023-00016-1]
- 36. Resistance of Gram-Negative Bacteria to Current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7144564/]
- 37. How to Combat Gram-Negative Bacteria Using Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8698890/]
- 38. Targeting bacterial membrane function - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3496266/]
- 39. Antimicrobial Peptides: Classification, Design, Application ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.582779/full]
- 40. Strategies to combat Gram-negative bacterial resistance ... [https://ophrp.org/journal/view.php?number=740]
- 41. A Foundation Model Identifies Broad-Spectrum ... [https://www.nature.com/articles/s41467-024-51933-2]
- 42. Combating Gram-negative infections: The role of ... [https://www.sciencedirect.com/science/article/pii/S2590006425009524]
- 43. Application of Polymeric Nanocarriers for Enhancing the ... [https://www.mdpi.com/2076-3417/11/22/10695]
- 44. Resistance mechanisms in Gram-negative bacteria [https://medintensiva.org/en-resistance-mechanisms-in-gram-negative-bacteria-articulo-S2173572722000947]
- 45. Antimicrobial peptide biological activity, delivery systems ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06321-9]
- 46. FAQs – Wissensplattform nanopartikel.info [https://nanopartikel.info/en/basics/faqs/]
- 47. Technical Nanomaterial FAQ's [https://cerionnano.com/2023/05/technical-faqs-product-integration-with-nanomaterials/]
- 48. Nanotechnology Safety Resources [https://www.acs.org/about/governance/committees/chemical-safety/publications-resources/nanotechnology-safety-resources.html]
- 49. Nanotechnology Frequently Asked Questions [https://www.cdc.gov/niosh/nano/faqs/index.html]
- 50. Overcoming Intrinsic and Acquired Resistance Mechanisms Associated with the Cell Wall of Gram-Negative Bacteria - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7558195/]
- 51. A synthetic lethal approach for compound and target ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4684722/]
- 52. Systems biology-guided identification of synthetic lethal ... [https://www.nature.com/articles/srep16025]
- 53. Natural Compounds With Antimicrobial and Antiviral Effect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8450524/]
- 54. Plant antibacterials: The challenges and opportunities [https://www.sciencedirect.com/science/article/pii/S2405844024071767]
- 55. Emerging antimicrobial therapies for Gram-negative ... [https://www.nature.com/articles/s44259-025-00087-2]
- 56. Drug combinations targeting antibiotic resistance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11721080/]
- 57. Synergistic combinations of antimicrobial peptides and ... [https://www.sciencedirect.com/science/article/pii/S0031699725075131]
- 58. Synergistic action of antimicrobial peptides and antibiotics [https://pmc.ncbi.nlm.nih.gov/articles/PMC11322369/]
- 59. New potentiators of ineffective antibiotics: Targeting the ... [https://www.sciencedirect.com/science/article/abs/pii/S1367593121001368]
- 60. High-throughput identification and rational design of ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.2001644]
- 61. CombiANT: Antibiotic interaction testing made easy [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000856]
- 62. Synergistic combination of two antimicrobial agents closing ... [https://www.nature.com/articles/s41598-018-25714-z]
- 63. Combating Gram-negative infections: The role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546895/]
- 64. Development and Challenges of Antimicrobial Peptides for ... [https://www.mdpi.com/2079-6382/9/1/24]
- 65. Mechanisms of Antimicrobial Peptide Resistance in Gram- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4410734/]
- 66. A How-To Guide for Mode of Action Analysis of Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7604286/]
- 67. Reports identify weakness in global pipeline for new ... [https://www.cidrap.umn.edu/antimicrobial-stewardship/reports-identify-weakness-global-pipeline-new-antibiotics-diagnostics]
- 68. 'A time of urgency': Crisis in the antibacterial pipeline [https://www.healio.com/news/infectious-disease/20251126/a-time-of-urgency-crisis-in-the-antibacterial-pipeline]
- 69. Antibiotics re-booted—time to kick back against drug ... [https://www.nature.com/articles/s44259-025-00096-1]
- 70. Antibiotic resistance: The challenges and some emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9459344/]
- 71. Strategies to combat Gram-negative bacterial resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10626324/]
- 72. IDSA 2024 Guidance on the Treatment of Antimicrobial ... [https://www.idsociety.org/practice-guideline/amr-guidance/]
- 73. Core Elements of Hospital Antibiotic Stewardship Programs [https://www.cdc.gov/antibiotic-use/hcp/core-elements/hospital.html]
- 74. Antimicrobial Stewardship Programs: A Review of ... [https://www.mdpi.com/2079-6382/11/3/378]
- 75. , adaptive and acquired antimicrobial Intrinsic in... resistance [https://pubmed.ncbi.nlm.nih.gov/28258229/]
- 76. To Push or To Pull? In a Post-COVID World, Supporting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7931625/]
- 77. Why pull incentives are key to catalysing private investment ... [https://efpia.eu/news-events/the-efpia-view/blog-articles/why-pull-incentives-are-key-to-catalysing-private-investment-into-antibiotics/]
- 78. Push and pull incentives [https://revive.gardp.org/resource/push-and-pull-incentives/?cf=encyclopaedia]
- 79. In vitro and ex vivo systems at the forefront of infection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7172914/]
- 80. Critical role of growth medium for detecting drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10936441/]
- 81. To overcome antibiotic resistance, new research says to let it ... [https://mcb.illinois.edu/news/2025-03-18/overcome-antibiotic-resistance-new-research-says-let-it-flow]
- 82. Intrinsic Resistance Patterns | MI - Microbiology [https://microbiology.mlsascp.com/intrinsic-resistance-patterns.html]
- 83. Impeding pathways of intrinsic resistance in Escherichia coli ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003443]
- 84. In vitro models for evaluating safety and efficacy of novel ... [https://pubmed.ncbi.nlm.nih.gov/27612408/]
- 85. Approach to combat antibiotic resistance turns bacterium's ... [https://www.stjude.org/media-resources/news-releases/2025-medicine-science-news/approach-to-combat-antibiotic-resistance-turns-bacteriums-genes-against-it.html]
- 86. Prediction of Antimicrobial Resistance in Gram-Negative ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.01013/full]
- 87. Building a Morbidostat: An automated continuous-culture ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3708598/]
- 88. Operation of morbidostat system [https://bio-protocol.org/exchange/minidetail?id=761864&type=30]
- 89. Morbidostat PID algorithm, tuning, and suggestions [https://forum.pioreactor.com/t/morbidostat-pid-algorithm-tuning-and-suggestions/116]
- 90. Shared and Unique Evolutionary Trajectories to ... [https://pubmed.ncbi.nlm.nih.gov/34154405/]
- 91. Experimental evolution in morbidostat reveals converging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8209735/]
- 92. Susceptibility and resistance of Gram-negative bacteria to ... [https://www.frontiersin.org/journals/antibiotics/articles/10.3389/frabi.2025.1615821/full]
- 93. Evaluation of Antibiotic Resistance Mechanisms in Gram ... [https://www.mdpi.com/2079-6382/12/11/1590]
- 94. High-throughput assessment of bacterial growth inhibition ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3182142/]
- 95. Troubleshooting Guide: Ensuring Optimal Performance [https://www.tmmedia.in/troubleshooting-guide-ensuring-optimal-performance/]
- 96. WHO: Reviews of antibacterial therapeutics and ... [https://amr.solutions/2025/10/04/who-reviews-of-antibacterial-therapeutics-and-diagnostics/]
- 97. Analysis of antibacterial agents in clinical and preclinical ... [https://www.who.int/publications/i/item/9789240113091]