Beyond the Hype: A Practical Guide to Training Robust Binding Affinity Models with PDBbind CleanSplit
Accurately predicting protein-ligand binding affinity is a cornerstone of computational drug discovery, yet the field has been hampered by overstated model performance due to pervasive data leakage in standard benchmarks.
Beyond the Hype: A Practical Guide to Training Robust Binding Affinity Models with PDBbind CleanSplit
Abstract
Accurately predicting protein-ligand binding affinity is a cornerstone of computational drug discovery, yet the field has been hampered by overstated model performance due to pervasive data leakage in standard benchmarks. This article provides a comprehensive guide for researchers and drug development professionals on the PDBbind CleanSplit dataset, a newly curated resource designed to eliminate train-test leakage and enable genuine assessment of model generalizability. We explore the foundational reasons for its development, detail methodological approaches for effective model training, address common troubleshooting and optimization challenges, and present a rigorous validation framework for comparing model performance. By adopting CleanSplit, the scientific community can build more reliable and trustworthy predictive models, ultimately accelerating the development of new therapeutics.
The Data Leakage Problem: Why PDBbind CleanSplit is a Necessity, Not an Option
The accurate prediction of protein-ligand binding affinity is a critical objective in structure-based drug design (SBDD). For years, the scientific community has relied on benchmarks derived from the PDBbind database and the Comparative Assessment of Scoring Functions (CASF) to evaluate the performance of novel computational models [1] [2]. However, a growing body of evidence reveals a fundamental flaw in this evaluation paradigm: widespread train-test data leakage has severely inflated performance metrics, creating an illusion of progress while masking poor generalization on truly novel complexes [1].
Data leakage occurs when information from outside the training dataset is used to create the model, leading to overly optimistic performance estimates that would not be achievable in real-world prediction scenarios [3] [4]. This issue is particularly pervasive in binding affinity prediction, where the standard practice of training on PDBbind and testing on CASF benchmarks has been compromised by undisclosed similarities between the training and test complexes [1] [2]. This review examines the sources and impacts of this leakage, presents a rigorous solution in the form of the PDBbind CleanSplit dataset, and provides protocols for developing leakage-free binding affinity models with genuinely generalizable performance.
The Data Leakage Problem in PDBbind-CASF
Mechanisms and Magnitude of Leakage
The data leakage between PDBbind and CASF benchmarks is not merely theoretical but stems from concrete structural similarities that enable models to "memorize" rather than "learn" true binding principles. A multimodal filtering algorithm analyzing protein similarity, ligand similarity, and binding conformation similarity has revealed alarming levels of contamination [1].
Table 1: Quantified Data Leakage Between PDBbind and CASF Benchmarks
| Similarity Metric | Threshold Value | Percentage of CASF Complexes Affected | Number of Similar Train-Test Pairs |
|---|---|---|---|
| Protein Similarity (TM-score) | >0.7 | 49% | ~600 |
| Ligand Similarity (Tanimoto) | >0.9 | Not specified | Significant |
| Binding Conformation (pocket-aligned RMSD) | Low values | Correlated with protein/ligand similarity | Part of the ~600 pairs |
The structural analysis demonstrates that nearly half of all CASF complexes share striking similarities with complexes in the PDBbind training set, complete with closely matched affinity labels [1]. This means models can achieve apparently state-of-the-art performance simply by recognizing structural patterns they encountered during training, rather than by genuinely understanding protein-ligand interactions.
Impact on Model Performance and Generalization
The consequences of this data leakage are profound. When state-of-the-art models like GenScore and Pafnucy were retrained on a leakage-free dataset (PDBbind CleanSplit), their performance on the CASF benchmark dropped substantially [1]. This performance collapse confirms that previously reported metrics were artificially inflated and did not reflect true generalization capability.
This phenomenon extends beyond structural bioinformatics. A systematic review found that data leakage has affected at least 294 scientific publications across 17 different scientific fields, potentially contributing to a broader reproducibility crisis in machine learning-based science [4]. In medical applications, models trained with leakage can fail catastrophically when deployed in real-world clinical settings, sometimes misclassifying most healthy patients as diseased when overt diagnostic features are removed [5].
Solution: PDBbind CleanSplit Protocol
Multimodal Filtering Algorithm
The PDBbind CleanSplit methodology employs a sophisticated structure-based clustering algorithm that simultaneously evaluates three dimensions of similarity to identify and remove problematic overlaps [1].
Table 2: Similarity Metrics in the CleanSplit Filtering Algorithm
| Metric | Measurement Target | Technical Implementation | Purpose in Leakage Prevention |
|---|---|---|---|
| TM-score | Protein structure similarity | Protein structure alignment | Eliminates test complexes with highly similar protein folds |
| Tanimoto coefficient | Ligand chemical similarity | Molecular fingerprint comparison | Removes training complexes with nearly identical ligands |
| Pocket-aligned RMSD | Binding conformation similarity | Ligand alignment within binding pocket | Filters complexes with similar binding modes |
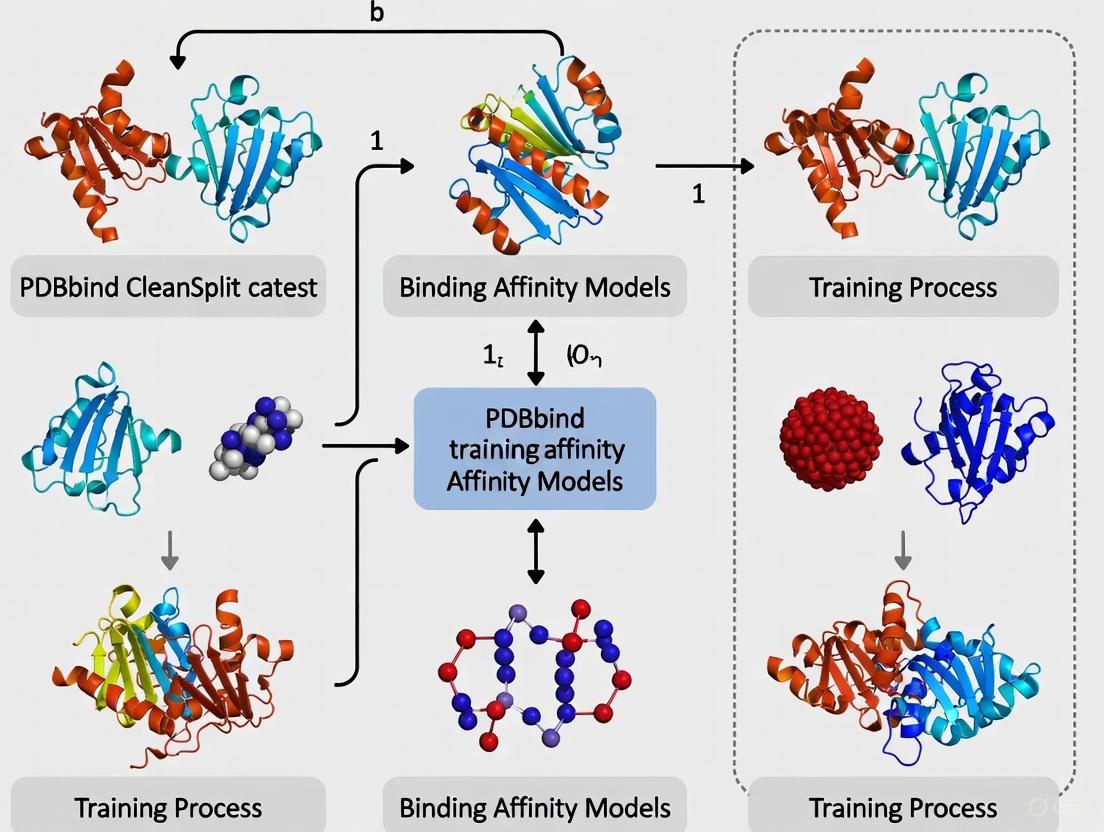
The filtering workflow operates iteratively, first addressing train-test leakage between PDBbind and CASF, then resolving redundancies within the training set itself. This process ultimately removes approximately 11.8% of training complexes (4% for direct train-test leakage and 7.8% for internal redundancies) [1].
The following diagram illustrates the comprehensive filtering workflow:
Independent Validation with BDB2020+
To enable truly external validation, researchers have created BDB2020+, an independent dataset constructed by matching high-quality binding free energies from BindingDB with co-crystalized ligand-protein complexes from the PDB deposited since 2020 [2]. This dataset is filtered using the same similarity control criteria as LP-PDBBind (a related leakage-proof dataset), ensuring no overlap with the training data and providing a rigorous testbed for model generalization [2].
Experimental Validation of the CleanSplit Approach
Performance Comparison on Standard Benchmarks
Retraining existing models on PDBbind CleanSplit provides a sobering reality check for the field. The following table compares benchmark performance before and after addressing data leakage:
Table 3: Model Performance With and Without Data Leakage
| Model | Original CASF Performance (with leakage) | Performance on CleanSplit (leakage-free) | Performance Change | Generalization to BDB2020+ |
|---|---|---|---|---|
| GenScore | High (original paper) | Substantially dropped | Significant decrease | Not specified |
| Pafnucy | High (original paper) | Substantially dropped | Significant decrease | Not specified |
| GEMS (GNN) | Not applicable | Maintains high performance | Minimal decrease | Good performance |
| IGN (retrained) | Not applicable | Improved compared to original | Increase | Better generalization [2] |
The performance degradation observed in models like GenScore and Pafnucy confirms that their original high performance was largely driven by data leakage rather than genuine learning of protein-ligand interactions [1]. In contrast, the Graph Neural Network for Efficient Molecular Scoring (GEMS) maintains high benchmark performance even when trained on CleanSplit, suggesting it possesses more robust generalization capabilities [1].
Ablation Study: Validating Learning Mechanisms
To confirm that model predictions are based on genuine understanding rather than spurious correlations, ablation studies are essential. When protein nodes were omitted from the GEMS graph architecture, the model failed to produce accurate predictions, confirming that its performance derives from actual protein-ligand interaction patterns rather than memorization of ligand structures alone [1].
This approach aligns with best practices for detecting data leakage, which include analyzing feature importance and verifying that models rely on logically relevant features rather than counter-intuitive proxies [3] [4].
Practical Implementation Protocols
Protocol 1: Creating a Leakage-Free Dataset Split
Purpose: To generate a customized leakage-free dataset for binding affinity prediction that ensures rigorous evaluation of model generalizability.
Materials:
- PDBbind general set (or other structural binding data)
- Structural similarity tools (TM-align for proteins, OpenBabel for ligand fingerprints)
- Clustering software (custom Python scripts)
Procedure:
- Protein Similarity Filtering:
- Calculate all-against-all TM-scores for proteins in candidate training and test sets
- Apply threshold: exclude training complexes with TM-score >0.7 to any test complex
- Retain only complexes with low structural similarity across splits
Ligand Similarity Filtering:
- Generate molecular fingerprints (ECFP4 or similar) for all ligands
- Calculate Tanimoto coefficients between all ligand pairs
- Apply threshold: exclude training complexes with Tanimoto >0.9 to any test ligand
Binding Conformation Validation:
- For remaining complexes, align binding pockets and calculate RMSD of ligand poses
- Manually inspect complexes with low RMSD values for similar binding modes
- Exclude training complexes with nearly identical binding conformations to test complexes
Internal Redundancy Reduction:
- Identify similarity clusters within the training set using the same metrics
- Iteratively remove complexes from each cluster until maximum within-cluster TM-score <0.8 and Tanimoto <0.85
- Preserve diverse representation across protein folds and ligand scaffolds
Validation: Confirm that no test complex has close analogs in training set using the defined similarity metrics. Verify dataset diversity through principal component analysis of ligand chemical space.
Protocol 2: Training a Robust Binding Affinity Model
Purpose: To train a graph neural network model for binding affinity prediction that generalizes to novel protein-ligand complexes.
Materials:
- PDBbind CleanSplit or LP-PDBBind dataset
- Graph neural network framework (PyTorch Geometric/DGL)
- Pretrained language models (ProtBERT for proteins, ChemBERTa for ligands)
- High-performance computing resources with GPU acceleration
Procedure:
- Data Preprocessing:
- Extract protein-ligand complexes from cleaned dataset
- Represent proteins as graphs: nodes as amino acids with structural features, edges based on spatial proximity
- Represent ligands as graphs: nodes as atoms with chemical features, edges as bonds
- Generate protein sequence embeddings using ProtBERT
- Generate ligand molecular embeddings using ChemBERTa
Model Architecture (GEMS-inspired):
- Implement sparse graph representation of protein-ligand interactions
- Incorporate pretrained embeddings as node features
- Use message-passing neural network layers with attention mechanisms
- Add rotational invariant spatial encoding for 3D structural information
- Include distance-aware aggregation functions
Training Protocol:
- Initialize model with transfer learning from language model embeddings
- Use robust loss function (Huber loss) for affinity prediction
- Apply regularization techniques: dropout, weight decay, early stopping
- Optimize using AdamW optimizer with learning rate scheduling
- Implement gradient clipping for training stability
Validation and Testing:
- Evaluate on CleanSplit test set using RMSE and Pearson correlation
- Test on completely independent dataset (BDB2020+)
- Perform ablation studies to verify protein contribution to predictions
- Compare against baseline models trained with standard splits
Expected Outcomes: Model should maintain performance on leakage-free test sets and show robust generalization to independent benchmarks like BDB2020+, with minimal performance gap between validation and external test sets.
Research Reagent Solutions
Table 4: Essential Tools for Leakage-Free Binding Affinity Prediction
| Resource | Type | Function | Application Notes |
|---|---|---|---|
| PDBbind CleanSplit | Dataset | Leakage-free training data for affinity prediction | Curated via multimodal filtering; strictly separated from CASF |
| LP-PDBBind [2] | Dataset | Reorganized PDBbind with minimal similarity between splits | Controls for protein sequence and ligand chemical similarity |
| BDB2020+ [2] | Benchmark | Independent validation set from post-2020 structures | True external test for generalization capability |
| TM-align [1] | Software tool | Protein structure similarity assessment | Used for calculating TM-scores in filtering algorithm |
| GEMS Framework [1] | Model architecture | Graph neural network with transfer learning | Maintains performance on leakage-free data |
| IGN (Interaction GraphNet) [2] | Model architecture | Graph neural network for protein-ligand structures | Recommended for scoring/ranking after retraining on LP-PDBBind |
| ProtBERT [6] | Pretrained model | Protein sequence representation | Provides transfer learning for protein encoding |
| ChemBERTa [6] | Pretrained model | Molecular representation from SMILES | Enables transfer learning for ligand encoding |
The following diagram illustrates the complete experimental workflow for developing and validating a leakage-free binding affinity prediction model:
The discovery of extensive train-test leakage between PDBbind and CASF benchmarks represents a critical inflection point for computational drug discovery. The field must transition from evaluating models on compromised benchmarks to adopting rigorous, leakage-free evaluation frameworks like PDBbind CleanSplit. The protocols and reagents outlined here provide a pathway for developing binding affinity models with genuinely generalizable performance, ultimately accelerating the identification of therapeutic candidates through more reliable computational predictions.
The accurate prediction of protein-ligand binding affinity is a cornerstone of computational drug design. For years, the field has relied on benchmarks that suggested continuous improvement in model performance. However, recent research has revealed a critical flaw in this narrative: widespread data leakage between the primary training dataset, PDBbind, and the standard evaluation benchmarks from the Comparative Assessment of Scoring Functions (CASF) has severely inflated performance metrics and led to an overestimation of model generalization capabilities [1] [2].
This data leakage occurs when models encounter test complexes that are highly similar to those seen during training, enabling prediction through memorization rather than learning fundamental principles of molecular recognition [1]. Alarmingly, some models maintain competitive benchmark performance even when critical structural information is omitted, suggesting they are not genuinely learning protein-ligand interactions [1] [7].
To address this fundamental challenge, we introduce PDBbind CleanSplit, a rigorously curated training dataset created via a novel structure-based filtering algorithm that eliminates train-test data leakage and reduces internal redundancies [1]. This application note provides a comprehensive overview of the CleanSplit methodology, validation protocols, and implementation guidelines to enable robust binding affinity prediction.
The Data Leakage Problem in PDBbind
The PDBbind database serves as the primary resource for training protein-ligand binding affinity prediction models. Its standard organization includes "general" and "refined" sets for training, with a separate "core" set used for testing, typically through the CASF benchmark [2]. This arrangement has been shown to contain significant data leakage, fundamentally compromising model evaluation.
Quantifying the Data Leakage
Analysis using structure-based clustering revealed extensive similarities between training and test complexes [1]:
| Similarity Type | Impact on CASF Complexes | Number of Similar Pairs |
|---|---|---|
| High structural similarity (Similar proteins, ligands, and binding conformation) | 49% of CASF complexes affected | Nearly 600 similarities identified |
| Ligand-based leakage (Tanimoto score > 0.9) | Additional data leakage pathway | Training complexes with identical ligands removed |
Consequences of Data Leakage
The impact of this data leakage on model performance evaluation is profound:
- Performance Inflation: Models achieve artificially high benchmark scores by memorizing structural similarities rather than learning generalizable interaction principles [1]
- Overestimated Generalization: Reported performance metrics do not reflect true capability on novel protein-ligand complexes [2]
- Impeded Progress: The field cannot accurately determine whether new scoring functions represent genuine improvements [2]
When state-of-the-art models like GenScore and Pafnucy were retrained on CleanSplit, their performance on the CASF benchmark dropped substantially, confirming that their previously reported high performance was largely driven by data leakage rather than true generalization capability [1].
The CleanSplit Methodology
PDBbind CleanSplit addresses data leakage through a multi-stage filtering approach that ensures strict separation between training and test complexes while simultaneously reducing redundancies within the training set.
Structure-Based Filtering Algorithm
The core innovation of CleanSplit is a structure-based clustering algorithm that performs multimodal similarity assessment between protein-ligand complexes. This algorithm evaluates three complementary dimensions of similarity:
Protein Similarity Assessment
- Metric: TM-score [1]
- Purpose: Quantifies protein structural similarity
- Advantage: More sensitive than sequence-based metrics, identifies structurally similar proteins even with low sequence identity [1]
Ligand Similarity Assessment
- Metric: Tanimoto score [1]
- Purpose: Quantifies chemical similarity between ligands
- Threshold: Training complexes with ligands having Tanimoto score > 0.9 to any CASF test ligand are removed [1]
Binding Conformation Assessment
- Metric: Pocket-aligned ligand root-mean-square deviation (RMSD) [1]
- Purpose: Quantifies similarity of ligand positioning within the protein binding pocket
- Significance: Ensures complexes with similar interaction patterns are identified
Redundancy Reduction within Training Set
Beyond addressing train-test leakage, CleanSplit also reduces internal redundancies within the training dataset:
- Problem: Approximately 50% of training complexes belong to similarity clusters [1]
- Solution: Iterative removal of complexes until striking similarity clusters are resolved [1]
- Result: 7.8% of training complexes removed to enhance dataset diversity [1]
This redundancy reduction discourages memorization and encourages learning of generalizable patterns, providing a more robust foundation for model training.
Experimental Validation
The effectiveness of PDBbind CleanSplit was validated through rigorous experimentation comparing model performance when trained on standard PDBbind versus the cleaned dataset.
Performance Comparison on CASF Benchmark
Retraining existing models on CleanSplit revealed their true generalization capabilities:
Table 1: Model Performance Comparison on CASF Benchmark
| Model | Performance Trained on Standard PDBbind | Performance Trained on CleanSplit | Performance Change |
|---|---|---|---|
| GenScore | High benchmark performance | Substantially dropped performance | Significant decrease |
| Pafnucy | High benchmark performance | Substantially dropped performance | Significant decrease |
| GEMS (Novel GNN) | Not applicable | Maintained high performance | State-of-the-art |
The GEMS Model: Maintaining Performance on CleanSplit
In contrast to existing models, the novel Graph Neural Network for Efficient Molecular Scoring (GEMS) maintained high benchmark performance when trained on CleanSplit, demonstrating genuine generalization capability [1]. Key architectural features include:
- Sparse Graph Modeling: Efficiently represents protein-ligand interactions [1]
- Transfer Learning: Leverages pre-trained language models [1]
- Ablation Study Validation: Model fails to produce accurate predictions when protein nodes are omitted, confirming predictions are based on genuine understanding of protein-ligand interactions [1]
Implementation Protocols
Dataset Acquisition and Preparation
Researchers can implement the CleanSplit methodology using the following protocol:
Table 2: Research Reagent Solutions for CleanSplit Implementation
| Resource | Type | Function in Protocol | Access Information |
|---|---|---|---|
| PDBbind Database | Data | Source of protein-ligand complexes and affinity data | http://www.pdbbind.org.cn/ [2] |
| CASF Benchmarks | Data | Evaluation datasets for generalization assessment | Included with PDBbind distribution |
| CleanSplit Filtering Algorithm | Software | Structure-based clustering and similarity assessment | Publicly available code [1] |
| Structural Biology Tools | Software | TM-score calculation, structural alignment | Publicly available (e.g., MMalign for TM-score) |
| Cheminformatics Toolkit | Software | Ligand similarity calculations (Tanimoto scores) | Open-source options (e.g., RDKit) |
Structural Data Preparation
- Download PDBbind general set (latest available version)
- Extract protein-ligand complexes and associated binding affinity data
- Standardize structures by adding missing hydrogen atoms and correcting bond orders [8] [9]
- Remove covalent complexes using CONECT record analysis [8] [9]
- Filter structures with steric clashes (heavy atom pairs < 2Å) [8] [9]
CleanSplit Filtering Protocol
The core filtering process follows these methodological steps:
Train-Test Separation Phase
- For each CASF test complex:
- Calculate TM-score against all PDBbind training complexes
- Compute Tanimoto scores for ligand pairs
- Determine pocket-aligned ligand RMSD for high-scoring pairs
- Identify and remove training complexes exceeding similarity thresholds:
- Verify separation: Ensure no high-similarity pairs remain between training and test sets
Internal Redundancy Reduction Phase
- Apply adapted filtering thresholds (slightly relaxed compared to train-test separation)
- Iteratively identify and resolve similarity clusters within training data
- Remove complexes until all striking similarity clusters are eliminated
- Preserve dataset size while maximizing diversity (balance between data quantity and quality)
Model Training and Evaluation Protocol
To ensure fair comparison and reproducible results:
Training Configuration:
- Train models on both standard PDBbind and CleanSplit for comparison
- Use identical hyperparameters and training procedures for both datasets
- Implement k-fold cross-validation with structure-based splitting
Evaluation Methodology:
- Assess performance on CASF benchmarks (2016 and later versions)
- Include additional independent test sets (e.g., BDB2020+ [2]) for generalization validation
- Report multiple metrics: Pearson R, RMSE, and ranking power
Ablation Studies:
- Test model performance with omitted protein or ligand information
- Validate that predictions require both protein and ligand inputs [1]
Integration with Broader Research Context
PDBbind CleanSplit represents part of a larger movement addressing data quality issues in computational drug discovery. Several related initiatives share similar goals:
Table 3: Related Data Curation Efforts in Binding Affinity Prediction
| Dataset/Approach | Primary Focus | Relationship to CleanSplit |
|---|---|---|
| LP-PDBBind [2] [10] | Minimize sequence and chemical similarity between splits | Complementary approach using different similarity metrics |
| HiQBind-WF [8] | Correct structural artifacts in protein-ligand complexes | Can be used as preprocessing step before CleanSplit filtering |
| PDBBind-Opt [9] | Automated workflow for structural preparation | Addresses complementary structural quality issues |
| Low Similarity Splits [11] | Minimize similarity leakage for benchmarking | Shared goal of improving generalization assessment |
These complementary approaches can be integrated into a comprehensive pipeline for preparing high-quality training data for binding affinity prediction.
PDBbind CleanSplit establishes a new standard for training and evaluating binding affinity prediction models. By addressing the critical issue of data leakage through rigorous structure-based filtering, it enables genuine assessment of model generalization capabilities. The substantial performance drop observed when existing models are retrained on CleanSplit reveals that previous benchmark results were largely driven by memorization rather than true learning of protein-ligand interactions.
The research community is encouraged to adopt CleanSplit as a benchmark for developing new scoring functions, particularly as the field advances toward more complex generative AI approaches for drug design [1]. Only through rigorous evaluation on truly independent test complexes can we develop models with genuine predictive power for novel drug targets.
Future directions include expanding the filtering approach to larger datasets, developing standardized benchmarking protocols, and integrating with structural quality improvement workflows to provide a comprehensive foundation for the next generation of binding affinity prediction models.
In the field of computational drug design, the accuracy of binding affinity predictions is paramount for effective structure-based drug design (SBDD). Benchmark datasets have long served as the gold standard for evaluating and advancing scoring functions. However, a critical issue has emerged: train-test data leakage between popular training sets and benchmark datasets has severely inflated performance metrics, leading to overestimation of model generalization capabilities [1] [12]. This application note examines how data similarity artificially boosts benchmark scores within the context of binding affinity prediction, focusing specifically on the PDBbind database and Comparative Assessment of Scoring Function (CASF) benchmarks. We present a detailed analysis of the leakage problem, quantify its effects, and provide validated protocols for creating leakage-free dataset splits using methods such as the PDBbind CleanSplit approach [1].
Quantitative Analysis of Data Leakage
Magnitude of Train-Test Similarity
Analysis using structure-based clustering algorithms has revealed substantial similarity between standard training datasets and evaluation benchmarks. The following table summarizes key quantitative findings from studies investigating the PDBbind-CASF relationship:
Table 1: Quantified Data Leakage Between PDBbind and CASF Benchmarks
| Metric | Value | Impact/Interpretation |
|---|---|---|
| Similar train-test pairs | Nearly 600 pairs | High structural similarity identified between PDBbind training and CASF test complexes [1] |
| Affected CASF complexes | 49% | Nearly half of benchmark complexes not presenting new challenges due to similarities [1] |
| Performance drop post-cleaning | Substantial | Retraining top models on cleaned data caused significant performance decreases [1] |
| Training set redundancy | ~50% of complexes | Approximately half of training complexes part of similarity clusters within training data [1] |
Impact on Model Performance
The consequences of this data leakage become evident when models are evaluated on properly cleaned datasets:
Table 2: Performance Impact of Data Leakage Removal
| Model/Training Condition | Performance Observation | Implication |
|---|---|---|
| State-of-the-art models (on original data) | Excellent benchmark performance | Overestimation of generalization capabilities [1] |
| Same models (on CleanSplit) | Marked performance drop | Previous performance largely driven by data leakage [1] |
| Graph Neural Network model (on CleanSplit) | Maintained high performance | Genuine generalization capability demonstrated [1] |
| Simple similarity-based algorithm | Competitive performance (R=0.716) | Performance achievable without understanding protein-ligand interactions [1] |
Detection Methodologies
Structure-Based Clustering Algorithm
The identification of data leakage requires a multi-modal approach to similarity assessment. The structure-based clustering algorithm proposed for creating PDBbind CleanSplit employs three complementary metrics [1]:
- Protein similarity: Quantified using TM-scores [1]
- Ligand similarity: Measured via Tanimoto scores [1]
- Binding conformation similarity: Calculated as pocket-aligned ligand root-mean-square deviation (r.m.s.d.) [1]
This combined approach robustly identifies complexes with similar interaction patterns, even when proteins share low sequence identity [1].
Data Leakage Root Causes
Understanding the fundamental causes of data leakage is essential for developing effective detection strategies:
- External Factors: Data acquisition methods and inherent data similarities can create leakage opportunities, particularly with correlated samples or repetitive content [13].
- Implementation Errors: Incorrect data splitting, improper data augmentation, and faulty synthetic data generation can introduce leakage during dataset preparation [13].
- Group Leakage: Multiple samples from the same source (e.g., same patient in medical imaging) distributed across training and test sets [13].
- Temporal Leakage: Time-series data split without regard to chronological order, training on future data to predict past events [13].
Figure 1: Workflow for detecting and mitigating data leakage in protein-ligand complex datasets using multi-modal similarity assessment.
Experimental Protocols
Protocol: Creating a Clean Dataset Split
This protocol describes the procedure for generating a leakage-free dataset based on the PDBbind CleanSplit methodology [1].
Materials and Equipment
- Hardware: Standard workstation with sufficient storage for structural datasets
- Software: Molecular visualization software, similarity calculation tools (TM-score, Tanimoto coefficients, RMSD calculation)
- Data: PDBbind database (or relevant structural dataset)
Procedure
Data Collection and Preprocessing
- Download the complete PDBbind dataset
- Standardize protein and ligand representations
- Resolve any missing residues or atoms
Similarity Calculation
- Compute all pairwise protein similarities using TM-score
- Calculate all pairwise ligand similarities using Tanimoto coefficients
- Determine binding conformation similarities using pocket-aligned ligand RMSD
Threshold Application
- Set similarity thresholds based on biological relevance
- Protein similarity threshold: Based on structural homology
- Ligand similarity threshold: Tanimoto > 0.9 for identifying nearly identical ligands [1]
- Binding conformation threshold: RMSD value indicating similar binding modes
Similarity Cluster Identification
- Identify complexes exceeding similarity thresholds
- Mark protein-ligand complexes sharing high similarity with benchmark sets
- Flag redundant complexes within the training set itself
Dataset Filtering
- Remove all training complexes closely resembling any test complex
- Eliminate training complexes with ligands identical to those in test set
- Resolve internal training set redundancies by removing similar complexes
Validation
- Verify cleaned dataset maintains sufficient size for training
- Ensure diverse representation of protein families and ligand types
- Confirm elimination of high-similarity pairs between training and test sets
Protocol: Evaluating Data Leakage in Existing Models
This protocol assesses whether a model's performance is inflated by data leakage.
Materials and Equipment
- Trained binding affinity prediction models
- Original and cleaned dataset splits
- Standard benchmarking environment
Procedure
Baseline Performance Establishment
- Evaluate model performance on standard benchmark using original training data
- Record key metrics (Pearson R, RMSD, etc.)
Retraining on Cleaned Data
- Retrain the same model architecture on the cleaned dataset split
- Maintain identical hyperparameters and training procedures
Performance Comparison
- Evaluate retrained model on the same benchmark
- Compare performance metrics with original model
Interpretation
- Significant performance drops suggest previous metrics were inflated by data leakage
- Maintained performance indicates genuine generalization capability
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| PDBbind CleanSplit | Leakage-free training dataset | Filtered using structure-based clustering; eliminates train-test similarity [1] |
| Structure-based clustering algorithm | Multi-modal similarity assessment | Combines protein, ligand, and binding conformation metrics [1] |
| Graph Neural Network (GEMS) | Binding affinity prediction | Maintains performance on cleaned data; sparse graph modeling [1] |
| LP-PDBBind | Alternative reorganized dataset | Controls for protein and ligand sequence/structural similarity [14] |
| TM-score | Protein structural similarity metric | Identifies similar protein folds beyond sequence identity [1] |
| Tanimoto coefficient | Ligand similarity metric | Quantifies 2D molecular similarity; threshold >0.9 for near-identical ligands [1] |
| Pocket-aligned ligand RMSD | Binding conformation similarity | Measures similar ligand positioning in protein binding sites [1] |
Figure 2: Framework for evaluating whether model performance is artificially inflated by data leakage or reflects genuine generalization capability.
The quantification of data leakage between the PDBbind database and CASF benchmarks reveals that nearly half of all test complexes share strong similarities with training data, significantly inflating perceived model performance [1]. The implementation of cleaned dataset splits such as PDBbind CleanSplit provides a necessary correction, enabling proper assessment of model generalization. The experimental protocols presented herein offer researchers practical methodologies for both creating leakage-free datasets and evaluating the true capabilities of binding affinity prediction models. As the field advances toward more reliable computational drug design, addressing data leakage systematically is essential for developing scoring functions with genuine predictive power for novel protein-ligand interactions.
The Impact of Dataset Redundancy on Model Training and Memorization
Dataset redundancy and data leakage represent critical, often overlooked, challenges in developing machine learning models for scientific applications, particularly in computational drug discovery. In the field of protein-ligand binding affinity prediction, these issues have led to widespread overestimation of model capabilities, with models learning to exploit statistical artifacts rather than underlying biological principles. The PDBbind database, a cornerstone resource for training scoring functions, has been shown to contain significant structural similarities and overlaps with standard benchmark sets like the Comparative Assessment of Scoring Functions (CASF). This redundancy creates a scenario where models can achieve impressive benchmark performance through memorization and pattern matching rather than genuine understanding of protein-ligand interactions [1] [15]. This application note examines the impact of dataset redundancy on model training, documents the creation of rigorously curated alternatives, and provides protocols for developing models that generalize to truly novel complexes.
The Data Redundancy Problem in Structural Biology
Quantifying Data Leakage in PDBbind
The standard practice of training on the PDBbind general set and evaluating on the CASF benchmark has been fundamentally compromised by data leakage. A rigorous structure-based analysis revealed alarming levels of similarity between training and test complexes:
Table 1: Quantified Data Leakage Between PDBbind and CASF Benchmarks
| Similarity Metric | Threshold Value | Percentage of CASF Complexes Affected | Impact on Model Performance |
|---|---|---|---|
| Overall Complex Similarity | TM-score, Tanimoto, & RMSD | 49% of CASF complexes had highly similar counterparts in training [1] | Enables near-direct label memorization |
| Ligand Similarity | Tanimoto > 0.9 | Significant number of ligands nearly identical between sets [1] | Models memorize ligand-affinity relationships |
| Protein Similarity | High TM-score | Structural similarities even with low sequence identity [1] | Exploitable through protein structure matching |
This leakage explains the paradoxical findings that some models maintain high performance even when critical input information (e.g., protein or ligand structures) is omitted, indicating they are not learning genuine interaction principles [15].
Memorization Versus Generalization
When models train on redundant datasets, they gravitate toward memorization-based shortcuts rather than learning the underlying relationship between structure and function. Studies systematically investigating these biases found that Atomic Convolutional Neural Network (ACNN) models performed comparably well on binding affinity prediction whether they were provided with full complex structures, ligand-only information, or protein-only information [15]. This clearly demonstrates that the models were leveraging dataset-specific biases rather than learning true structure-activity relationships.
Protocols for Creating Non-Redundant Datasets
The PDBbind CleanSplit Framework
The PDBbind CleanSplit methodology establishes a new standard for creating training datasets with minimized redundancy and data leakage [1]. The protocol employs a structure-based clustering algorithm that performs multimodal filtering based on three key similarity metrics:
- Protein Similarity: Calculated using TM-scores [1]
- Ligand Similarity: Calculated using Tanimoto scores [1]
- Binding Conformation Similarity: Calculated using pocket-aligned ligand root-mean-square deviation (RMSD) [1]
Protocol: Implementing the CleanSplit Filtering Algorithm
- Input: PDBbind general set (training) and CASF core sets (test)
- Step 1 - Cross-Set Filtering: Identify and remove all training complexes with:
- Combined protein, ligand, and binding pose similarity above threshold to any test complex
- Ligands with Tanimoto similarity >0.9 to any test ligand
- Step 2 - Intra-Set Deduplication: Iteratively identify similarity clusters within training data using adapted thresholds and remove complexes until all striking clusters are resolved
- Output: PDBbind CleanSplit training set (4% reduction from original) with strict separation from test benchmarks
The following workflow diagram illustrates the CleanSplit creation process:
Complementary Data Quality Improvements
Concurrent efforts address additional data quality issues in PDBbind that further hamper model generalizability. The HiQBind workflow applies systematic structural corrections through several automated modules [16] [17]:
Protocol: HiQBind-WF Structural Correction Steps
- Input: Raw PDB files from PDBbind or other sources
- Step 1 - Filtering: Apply successive filters to remove:
- Covalent binders (using CONECT records)
- Ligands with rare elements (beyond H, C, N, O, F, P, S, Cl, Br, I)
- Very small ligands (<4 heavy atoms)
- Complexes with severe steric clashes (<2Å heavy atom distances)
- Step 2 - Ligand Fixing: Correct bond orders, protonation states, and aromaticity
- Step 3 - Protein Fixing: Add missing atoms and residues in binding site regions
- Step 4 - Structure Refinement: Add hydrogens to the protein-ligand complex state (rather than separately)
- Output: High-quality, structurally plausible complexes for training
Experimental Validation of Redundancy Impact
Benchmarking Performance Drops with CleanSplit
Retraining existing models on PDBbind CleanSplit provides striking evidence of how data leakage had inflated reported performance metrics:
Table 2: Model Performance Comparison on Original vs. CleanSplit Training Data
| Model | Architecture Type | Performance on Original PDBbind | Performance on CleanSplit | Performance Drop |
|---|---|---|---|---|
| GenScore | Graph Neural Network | High benchmark performance (R² ~0.7 range) | Substantially reduced performance [1] | Up to 40% drop in R² score [18] |
| Pafnucy | 3D Convolutional Neural Network | High benchmark performance (R² ~0.49-0.73) [15] | Substantially reduced performance [1] | Significant drop (exact value not specified) [1] |
| Simple Search Algorithm | k-NN style similarity matching | Competitive with deep learning models [1] | N/A (demonstrates leakage mechanism) | Highlights memorization potential [1] |
The GEMS Model: Generalization Through Improved Architecture
To address the generalization challenge exposed by CleanSplit, the Graph neural network for Efficient Molecular Scoring (GEMS) was developed with specific architectural innovations:
Key Features of the GEMS Architecture:
- Sparse Graph Modeling: Represents protein-ligand interactions as sparse graphs rather than dense grids, improving efficiency and generalization [1]
- Transfer Learning Integration: Leverages pre-trained language models (ESM2 for proteins, ChemBERTa for ligands) to incorporate evolutionary and chemical information [1] [18]
- Ablation-Validated Learning: Performance collapses when protein nodes are removed, confirming genuine interaction learning rather than ligand memorization [1] [18]
When trained on CleanSplit, GEMS maintains high CASF benchmark performance where previous models show significant drops, demonstrating true generalization rather than data exploitation [1].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Resources for Robust Binding Affinity Model Development
| Resource Name | Type | Primary Function | Key Features |
|---|---|---|---|
| PDBbind CleanSplit | Curated Dataset | Training set with minimized redundancy | Strict separation from CASF benchmarks; reduced internal redundancy [1] |
| HiQBind-WF | Computational Workflow | Structural correction of protein-ligand complexes | Automated fixing of bonds, protonation, clashes; open-source [16] [17] |
| GEMS | Graph Neural Network | Binding affinity prediction | Sparse graph modeling; transfer learning integration; demonstrated generalization [1] [18] |
| DecoyDB | Pre-training Dataset | Self-supervised learning for complexes | 61K ground truth + 5.3M decoy structures; enables contrastive pre-training [19] |
| CASF Benchmark | Evaluation Suite | Standardized model assessment | Scoring, ranking, docking, and screening power metrics [1] [16] |
Future Directions and Implementation Recommendations
The field is moving toward more rigorous training paradigms to combat dataset redundancy:
- Self-Supervised Learning: DecoyDB enables contrastive pre-training on large-scale unlabeled complexes before fine-tuning on limited affinity data, improving data efficiency [19]
- Structural Quality Focus: HiQBind and similar initiatives address fundamental data quality issues beyond redundancy [16] [17]
- Ablation Testing: Rigorous validation should include controls that remove key inputs (e.g., protein structure) to confirm models learn genuine interactions [1] [15]
Implementation Protocol for Model Development:
- Step 1: Start with structurally validated datasets (HiQBind-WF processed)
- Step 2: Apply rigorous data splits (CleanSplit methodology) to ensure test independence
- Step 3: Consider pre-training on decoy datasets (DecoyDB) when labeled data is limited
- Step 4: Employ architectures with structural and biochemical priors (GEMS-style sparse graphs)
- Step 5: Validate with ablation studies to confirm learning of genuine protein-ligand interactions
The recognition of dataset redundancy as a critical factor in binding affinity prediction represents a maturation of the field. By adopting these curated datasets, rigorous protocols, and validated architectures, researchers can develop models with genuine generalization capability, ultimately accelerating robust drug discovery.
Building on a Clean Slate: Methodologies and Architectures for CleanSplit Training
Accurate prediction of protein-ligand binding affinity is a cornerstone of modern computational drug discovery, enabling researchers to identify promising therapeutic candidates more efficiently. The performance of machine learning models in this domain heavily depends on both the architectural choices and the quality of the training data. Historically, many models have been trained on benchmark datasets like PDBBind, but emerging research reveals that conventional data splitting methods can introduce significant data leakage, compromising model generalizability. Data leakage occurs when highly similar proteins or ligands appear in both training and testing sets, leading to artificially inflated performance metrics that do not reflect true predictive capability on novel complexes [10]. This application note examines the integration of three prominent neural network architectures—Graph Neural Networks (GNNs), Convolutional Neural Networks (CNNs), and Transformers—with the rigorously curated LP-PDBBind (Leak-Proof PDBBind) dataset. We provide a detailed comparative analysis and experimental protocols to guide researchers in developing more generalizable and reliable binding affinity models, framed within the broader thesis that data cleanliness is equally as critical as model architecture for success in real-world drug discovery applications.
The LP-PDBBind Dataset: A Foundation for Generalizable Models
The standard PDBBind dataset is a widely used resource containing protein-ligand complexes and their experimentally measured binding affinities. However, its standard "general," "refined," and "core" sets are cross-contaminated with proteins and ligands of high sequence and structural similarity. This overlap means that models evaluated on the standard core set are often tested on data very similar to their training sets, rather than on truly novel complexes [2]. The LP-PDBBind dataset was created specifically to address this fundamental flaw.
LP-PDBBind reorganizes the PDBBind data through a meticulous splitting procedure that minimizes sequence and chemical similarity between the training, validation, and test datasets. This process involves:
- Similarity Control: Implementing stringent controls on both protein sequence similarity and ligand chemical similarity across dataset splits to prevent memorization and overfitting [10] [2].
- Data Cleaning: Removing covalently bound ligand-protein complexes to focus on non-covalent binding, filtering out ligands with rare atomic elements, and correcting inconsistencies in reported binding free energies and units [2] [20].
- Stratified Splitting: Creating data splits that ensure the protein-ligand structural interaction patterns are distinct among the training, validation, and test sets, providing a more challenging and realistic benchmark [2].
Retraining models on LP-PDBBind leads to more accurate assessments of their capabilities. While performance on the standard PDBBind test set may drop due to the removal of data leakage, the models demonstrate superior generalizability on truly independent test sets like BDB2020+, which is compiled from recent BindingDB entries and filtered with the same similarity criteria [10]. This makes LP-PDBBind an essential resource for developing scoring functions that perform reliably in prospective drug discovery campaigns.
Key Architectures for Binding Affinity Prediction
Table 1: Core Architectures for Protein-Ligand Binding Affinity Prediction
| Architecture | Core Input Representation | Key Strengths | Inherent Limitations |
|---|---|---|---|
| Convolutional Neural Networks (CNNs) | 3D voxelized grid of the binding pocket, with channels representing different atom types or chemical features [21]. | - Excels at extracting spatially local patterns and interactions.- Directly models the 3D structural environment.- Proven success in pose prediction and virtual screening [21]. | - Computationally intensive due to 3D convolutions.- Limited explicit modeling of long-range interactions or graph-structured data.- Resolution of the grid can impact performance. |
| Graph Neural Networks (GNNs) | Molecular graphs where nodes represent atoms (with features like type, charge) and edges represent bonds or distances [22]. | - Naturally represents molecular topology and non-Euclidean data.- Captures both local and global dependencies through message passing.- Models such as IGN show strong performance on cleaned datasets [10]. | - Performance can be sensitive to the definition of nodes, edges, and their features.- May require sophisticated architectures to capture complex 3D geometric relationships. |
| Transformers | Sequences (e.g., amino acid sequences, SMILES strings) or tokenized structural representations [23] [24]. | - Powerful attention mechanism captures long-range, global dependencies within and between sequences.- Can integrate information from multiple modalities (sequence, structure).- Enables prediction of conformational changes and population shifts [24]. | - High computational demand and data requirements for effective training.- Less intuitive for direct spatial reasoning compared to CNNs and GNNs. |
Quantitative Performance Benchmarking
The true efficacy of an architecture is revealed through its performance on leak-proof datasets. The following table summarizes benchmark results for various architectures retrained on the LP-PDBBind dataset and evaluated on independent test sets.
Table 2: Performance Benchmark of Architectures on Clean and Independent Datasets
| Model/Architecture | LP-PDBBind Test Set (Performance Metric) | BDB2020+ Independent Set (Performance Metric) | Key Application Context |
|---|---|---|---|
| InteractionGraphNet (IGN) [10] | Improved performance post-retraining | Significant improvement in generalizability | Scoring and ranking new protein-ligand systems [10] |
| GNNSeq [22] | Pearson Correlation Coefficient (PCC): ~0.784 (on PDBBind v.2020 refined set) | N/A | Sequence-based prediction; Virtual screening (AUC: 0.74 on DUDE-Z) |
| Ligand-Transformer [24] | Comparably better correlation on PDBBind2020 | N/A | Predicts affinity & conformational space; Hit identification (58% hit rate vs. EGFRLTC) |
| CNN (3D Grid-Based) [21] | Outperformed AutoDock Vina in pose ranking and virtual screening (on its test sets) | N/A | Pose prediction and virtual screening using 3D structural data |
Model Development on Clean Data
Detailed Experimental Protocols
Protocol 1: Training a GNN on LP-PDBBind for Binding Affinity Prediction
This protocol details the process for training a Graph Neural Network, specifically using the InteractionGraphNet (IGN) architecture, on the LP-PDBBind dataset to achieve robust binding affinity prediction.
- Objective: To develop a GNN-based scoring function that generalizes effectively to novel protein-ligand complexes by leveraging the leak-proof data splits of LP-PDBBind.
Research Reagent Solutions:
- LP-PDBBind Dataset: The foundational cleaned dataset with predefined train/validation/test splits, ensuring minimal similarity leakage [20].
- IGN Model Code: Implementation of the InteractionGraphNet, which uses GNNs to represent raw 3D protein and ligand structures [10].
- RDKit: Open-source cheminformatics toolkit used for processing ligand structures, calculating molecular descriptors, and handling SMILE strings [22] [20].
- PyTor Geometric (PyG): A library for deep learning on irregularly structured input data such as graphs, providing the core infrastructure for GNN implementation.
Step-by-Step Workflow:
- Data Acquisition and Preparation:
- Download the LP-PDBBind meta-information file (
LP_PDBBind.csv) from the THGLab GitHub repository [20]. - Obtain the corresponding protein (PDB) and ligand (SDF/MOL2) structure files from the official PDBBind website, using the PDB IDs listed in the meta-file.
- Filter the dataset for non-covalent binders by selecting only entries where the
covalentcolumn isFALSEand the desired clean level (e.g.,CL1) isTRUE[20].
- Download the LP-PDBBind meta-information file (
- Feature Engineering and Graph Construction:
- For each protein-ligand complex, represent the ligand as a molecular graph where nodes are atoms and edges are bonds or interatomic distances within a cutoff.
- Extract atom-level features for ligand atoms (e.g., atom type, hybridization, partial charge) and protein atoms (e.g., amino acid type, residue identity).
- Construct a heterogeneous graph that connects the ligand subgraph to nearby protein residues, defining edges based on spatial proximity to capture intermolecular interactions.
- Model Training and Optimization:
- Initialize the IGN model architecture, which processes the graph to learn a representation for the complex and outputs a binding affinity score.
- Train the model using the predefined LP-PDBBind training set, employing a regression loss function like Mean Squared Error (MSE) between predicted and experimental binding affinities (e.g., pKd).
- Use the LP-PDBBind validation set for hyperparameter tuning and early stopping to prevent overfitting.
- Model Evaluation:
- Perform the primary evaluation on the held-out LP-PDBBind test set to measure performance in a leak-proof setting.
- For a true test of generalizability, benchmark the final model on the independent BDB2020+ dataset [10] [2]. Report key metrics such as Pearson Correlation Coefficient (PCC), Root Mean Square Error (RMSE), and Mean Absolute Error (MAE).
- Data Acquisition and Preparation:
GNN Training Workflow
Protocol 2: Implementing a Sequence-Based Transformer for Virtual Screening
This protocol outlines the use of a Transformer model, such as Ligand-Transformer, for sequence-based virtual screening, which can be particularly powerful when structural data is limited or for large-scale screening.
- Objective: To employ a Transformer architecture for predicting protein-ligand binding affinity using primarily sequence information, enabling the screening of large compound libraries against target proteins.
Research Reagent Solutions:
- Ligand-Transformer Framework: A deep learning method based on the transformer architecture that takes the amino acid sequence of the target protein and the topology of the small molecule as input [24].
- BindingDB or TargetMol: Public and commercial databases of experimental binding data and purchasable compounds for sourcing active and decoy molecules for screening [24].
- GraphMVP Framework: Used within Ligand-Transformer to generate informed ligand representations by injecting knowledge of 3D molecular geometry into a 2D molecular graph encoder [24].
Step-by-Step Workflow:
- Data Preprocessing and Encoding:
- For the target protein, input the amino acid sequence. For the ligand, input a representation of its topology, such as a SMILES string or a 2D molecular graph.
- Tokenize the protein sequence and the ligand representation (e.g., using a chemical vocabulary for SMILES).
- Use pre-trained encoders (e.g., from AlphaFold for the protein and GraphMVP for the ligand) to generate high-dimensional initial representations for both molecules, capturing their intrinsic structural and chemical properties [24].
- Model Architecture and Fine-Tuning:
- Process the protein and ligand representations through a cross-modal attention network. This allows the model to exchange information between the protein and ligand representations, effectively "reasoning" about their potential interaction [24].
- The architecture typically includes downstream prediction heads for binding affinity (e.g., pKd) and optionally for other properties, such as interatomic distances.
- If a sufficiently large, task-specific dataset is available (e.g., EGFRLTC-290), fine-tune the pre-trained Ligand-Transformer model on this data to enhance its predictive accuracy for the specific target [24].
- Virtual Screening Execution:
- Apply the trained/fine-tuned model to a large library of compounds (e.g., the TargetMol subset used in the Ligand-Transformer study [24]).
- Rank the compounds based on their predicted binding affinity (e.g., lowest IC50 or highest pKd).
- Apply additional criteria based on the model's outputs, such as consistency across ensemble models or predicted binding mode characteristics, to select a shortlist of top candidates for experimental validation [24].
- Experimental Validation:
- Procure the top-ranked compounds and test them experimentally using binding assays (e.g., measuring IC50 values) to validate the model's predictions and identify true hits.
- Data Preprocessing and Encoding:
Integrated Discussion and Architectural Selection Guide
The choice of architecture is not a one-size-fits-all decision but should be guided by the specific research question, data availability, and application context. When working with the LP-PDBBind dataset, the following integrated considerations emerge:
- For Direct 3D Structure Utilization: If high-quality, co-crystalized protein-ligand structures are available and the primary goal is to leverage explicit 3D spatial information, CNN-based models are a strong choice. Their ability to learn from voxelized representations of the binding pocket makes them exceptionally suited for tasks like pose prediction and scoring when the 3D complex is known or can be reliably docked [21].
- For Robust Representation of Molecular Topology: If the research aims to balance structural information with robustness to conformational changes, or requires a natural representation of molecular connectivity, GNNs like IGN are highly recommended. Their performance on the LP-PDBBind benchmark and subsequent strong generalizability to new systems like those in BDB2020+ make them a top contender for robust scoring and ranking applications [10]. They are particularly powerful when the input is a molecular graph derived from a 3D structure.
- For Sequence-Based Screening and Leveraging Pre-trained Models: When 3D structural data is unavailable, unreliable, or when screening vast libraries against a target using only its amino acid sequence, Transformer models like Ligand-Transformer offer a powerful and flexible solution. Their ability to capture long-range dependencies and integrate information from sequences and ligand topologies enables them to predict not only affinity but also aspects of the bound conformational landscape, providing deeper mechanistic insight [24]. They are ideal for large-scale virtual screening in the absence of explicit structural complexes.
Ultimately, the most profound insight from recent research is that the careful curation of training data, as embodied by the LP-PDBBind dataset, is a force multiplier for any architectural choice. A simpler model trained on a rigorously leak-proof dataset can often generalize more effectively than a complex model trained on a contaminated benchmark. Therefore, the architectural selection should be made in concert with a commitment to utilizing the highest-quality, most generalizable data available.
Leveraging Pre-trained Models and Transfer Learning for Enhanced Feature Extraction
The field of computational drug design relies on accurate scoring functions to predict protein-ligand binding affinities, a critical task for structure-based drug design (SBDD). For years, the standard practice has involved training deep learning models on the PDBbind database and evaluating their generalization capability using the Comparative Assessment of Scoring Functions (CASF) benchmark datasets. However, recent research has exposed a fundamental flaw in this paradigm: widespread train-test data leakage between these datasets has severely inflated performance metrics, leading to overestimation of model generalization capabilities [25].
The groundbreaking PDBbind CleanSplit study revealed that nearly 49% of all CASF complexes had exceptionally similar counterparts in the training data, sharing not only similar ligand and protein structures but also comparable ligand positioning within protein pockets and closely matched affinity labels [25]. This redundancy enabled models to achieve high benchmark performance through simple memorization rather than genuine understanding of protein-ligand interactions. Alarmingly, some models performed comparably well on CASF benchmarks even after omitting all protein or ligand information from their input data, confirming they were not learning fundamental interaction principles [25] [26].
This data leakage crisis necessitates a fundamental shift in approach. This Application Note provides detailed protocols for leveraging pre-trained models and transfer learning to build robust binding affinity predictors that generalize effectively to novel protein-ligand complexes when trained on rigorously curated datasets like PDBbind CleanSplit.
The PDBbind CleanSplit Solution: A New Foundation for Model Training
The CleanSplit Filtering Methodology
PDBbind CleanSplit was created using a novel structure-based filtering algorithm that eliminates data leakage and reduces internal redundancies through a multi-stage process [25]:
- Structure-based clustering: Similarity between protein-ligand complexes is computed using a combined assessment of protein similarity (TM scores), ligand similarity (Tanimoto scores), and binding conformation similarity (pocket-aligned ligand root-mean-square deviation)
- Train-test separation: All training complexes closely resembling any CASF test complex are excluded, along with training complexes containing ligands identical to those in the test set (Tanimoto > 0.9)
- Redundancy reduction: Similarity clusters within the training dataset itself are identified and resolved using adapted filtering thresholds, removing 7.8% of training complexes to enhance diversity
Impact on Model Performance Assessment
Retraining existing top-performing models on CleanSplit caused substantial performance drops on benchmark tests, confirming their previous high scores were largely driven by data memorization [25]. This establishes CleanSplit as a more reliable foundation for developing truly generalizable binding affinity prediction models.
Table 1: Performance Impact of PDBbind CleanSplit on Existing Models
| Model Type | Performance on Original PDBbind | Performance on CleanSplit | Interpretation |
|---|---|---|---|
| GenScore | High benchmark performance | Substantially reduced performance | Previous performance inflated by data leakage |
| Pafnucy | High benchmark performance | Substantially reduced performance | Previous performance inflated by data leakage |
| Simple similarity-based algorithm | Competitive performance (Pearson R = 0.716) | N/A | Confirms benchmarks can be gamed through memorization |
Transfer Learning Framework for Binding Affinity Prediction
Meta-Learning Enhanced Transfer Learning Protocol
The following protocol combines meta-learning with transfer learning to mitigate negative transfer—a phenomenon where knowledge from source domains negatively impacts target task performance [27].
Phase 1: Source Domain Pre-processing
- Step 1: Collect abundant source domain data (e.g., protein kinase inhibitors from ChEMBL and BindingDB)
- Step 2: Standardize molecular representations (ECFP4 fingerprints, 4096 bits)
- Step 3: Transform affinity data to binary classification (active/inactive using threshold of 1000 nM)
- Step 4: Apply structural clustering to identify representative subsets
Phase 2: Meta-Learning for Sample Weighting
- Step 5: Define meta-model architecture with parameters φ
- Step 6: Initialize base model with parameters θ
- Step 7: For each iteration:
- Meta-model predicts weights for source data points
- Base model trains on weighted source data
- Base model predicts on target validation set
- Validation loss updates meta-model parameters
- Step 8: Output optimal source sample weights and base model initialization
Phase 3: Transfer Learning Execution
- Step 9: Pre-train base model on optimally weighted source data
- Step 10: Fine-tune on target domain (PDDBind CleanSplit)
- Step 11: Evaluate on strictly independent test sets (CASF benchmarks)
GEMS Architecture: Integrating Transfer Learning and Sparse Graph Modeling
The Graph Neural Network for Efficient Molecular Scoring (GEMS) demonstrates how transfer learning principles can be successfully applied within the CleanSplit framework [25]:
Architecture Components:
- Sparse graph modeling: Represents protein-ligand interactions as graphs with minimal redundant connections
- Transfer learning from language models: Leverages pre-trained protein language models for enhanced feature extraction
- Multi-scale feature integration: Combines atomic, residue, and molecular-level features
Implementation Protocol:
- Step 1: Initialize protein feature extractor with weights from pre-trained language model (e.g., ProtBERT, ESM)
- Step 2: Construct graph representation with:
- Nodes: Protein residues and ligand atoms
- Edges: Physicochemical interactions within cutoff distance
- Step 3: Implement message-passing layers with attention mechanisms
- Step 4: Train on PDBbind CleanSplit with standard affinity prediction loss function
- Step 5: Validate on strictly independent CASF benchmarks
Research Reagent Solutions for Transfer Learning Implementation
Table 2: Essential Research Reagents and Computational Tools
| Reagent/Tool | Type | Function in Protocol | Implementation Notes |
|---|---|---|---|
| PDBbind CleanSplit | Curated Dataset | Primary training data | Provides leakage-free foundation for model development |
| CASF 2016/2020 | Benchmark Dataset | Model evaluation | Strictly independent test sets for generalization assessment |
| ECFP4 Fingerprints | Molecular Representation | Compound structure encoding | 4096-bit fixed length, RDKit implementation |
| Protein Language Models (ESM, ProtBERT) | Pre-trained Models | Feature extraction initialization | Transfer learned protein representations |
| GEMS Architecture | Graph Neural Network | Binding affinity prediction | Sparse graph modeling of interactions |
| Meta-Weight-Net Algorithm | Meta-Learning Framework | Sample weighting optimization | Mitigates negative transfer between domains |
| RF-Score Features | Traditional ML Features | Baseline comparison | Atom-pair distance counts for random forest models |
| HiQBind-WF | Quality Control Workflow | Data preprocessing and validation | Corrects structural artifacts in protein-ligand complexes |
Experimental Protocols and Validation Methodologies
Protocol: Negative Transfer Mitigation in Kinase Inhibitor Prediction
This protocol demonstrates the meta-learning framework for predicting protein kinase inhibitors while mitigating negative transfer [27].
Materials:
- Kinase inhibitor data from ChEMBL (version 34) and BindingDB
- 7098 unique PKIs with activity against 162 protein kinases
- RDKit for molecular representation
- Meta-learning framework with base model and meta-model
Procedure:
- Data Curation:
- Filter to include only Ki values with molecular mass < 1000 Da
- Standardize structures and generate canonical nonisomeric SMILES strings
- Calculate geometric mean for multiple Ki values per compound
- Transform Ki values to binary classification (active/inactive using 1000 nM threshold)
Domain Specification:
- Define target data set:
T^(t) = {(x_i^t, y_i^t, s^t)}(inhibitors of data-reduced PK) - Define source data set:
S^(-t) = {(x_j^k, y_j^k, s^k)}_(k≠t)(PKIs of multiple PKs excluding target)
- Define target data set:
Model Definition:
- Base model
fwith parametersθfor classifying active/inactive compounds - Meta-model
gwith parametersφfor predicting sample weights
- Base model
Meta-Training Loop:
- For each iteration:
- Meta-model predicts weights for source data points
- Base model trains on weighted source data using weighted loss function
- Base model predicts on target training data
- Validation loss calculated and used to update meta-model parameters
- For each iteration:
Transfer Learning Execution:
- Pre-train base model using optimal weights from meta-learning
- Fine-tune on reduced target data
- Evaluate generalization on held-out test set
Validation:
- Compare against baseline models without meta-learning
- Statistical significance testing of performance differences
- Ablation studies on meta-learning components
Protocol: Binding Affinity Prediction with GEMS on CleanSplit
This protocol details the implementation of the GEMS model trained on PDBbind CleanSplit for binding affinity prediction [25].
Materials:
- PDBbind CleanSplit training dataset
- CASF 2016 and 2020 benchmark sets
- Graph neural network framework (PyTorch Geometric/DGL)
- Pre-trained protein language models
Procedure:
- Data Preparation:
- Load PDBbind CleanSplit training complexes
- Extract protein sequences and ligand structures
- Generate graph representations with:
- Protein residue nodes (features from language model)
- Ligand atom nodes (chemical features)
- Edges based on spatial proximity and interaction types
Model Initialization:
- Initialize protein feature extractor with pre-trained language model weights
- Initialize graph neural network with sparse attention mechanisms
- Set optimization parameters (learning rate, batch size, epochs)
Training Loop:
- For each batch of protein-ligand complexes:
- Construct graph representation
- Forward pass through GEMS architecture
- Calculate loss between predicted and experimental binding affinities
- Backward pass and parameter updates
- For each batch of protein-ligand complexes:
Validation:
- Evaluate on CASF benchmarks after each epoch
- Monitor for overfitting despite reduced leakage
- Conduct ablation studies (e.g., omitting protein nodes to test for genuine learning)
The integration of pre-trained models and transfer learning with rigorously curated datasets like PDBbind CleanSplit represents a paradigm shift in binding affinity prediction. The protocols outlined in this Application Note provide researchers with practical methodologies for developing models that generalize to novel protein-ligand complexes rather than merely memorizing training data.
Future directions in this field include:
- Expansion of the "smarter data" approach combining AI-generated synthetic structures with rigorous quality filtering [28]
- Integration of molecular dynamics simulations to capture conformational dynamics beyond static snapshots
- Participation in initiatives like Target2035 that aim to create massive, high-quality, standardized protein-ligand binding datasets [28]
- Development of more sophisticated meta-learning frameworks that dynamically balance positive and negative transfer across diverse biological targets
By adopting these protocols and contributing to the ongoing refinement of data curation and transfer learning methodologies, researchers can accelerate progress toward truly predictive computational drug design.
The accurate prediction of protein-ligand binding affinity is a fundamental challenge in computational drug design. Traditional scoring functions have shown limited accuracy, prompting the development of deep-learning-based alternatives [1]. However, a critical issue has undermined confidence in these new models: train-test data leakage between the primary training database (PDBbind) and standard evaluation benchmarks (CASF) [1] [2]. This leakage has artificially inflated performance metrics, leading to overestimation of model generalization capabilities [1].
This case study examines the implementation of a novel graph neural network model (GEMS) trained on PDBbind CleanSplit, a rigorously filtered dataset designed to eliminate data leakage and redundancy [1]. We present comprehensive application notes and experimental protocols for reproducing this approach, which demonstrates robust generalization to strictly independent test datasets through sparse graph modeling and transfer learning from language models [1].
Background and Problem Formulation
The Data Leakage Problem in PDBbind
The PDBbind database has served as the primary training resource for most scoring functions, with evaluation typically performed using the Comparative Assessment of Scoring Function (CASF) benchmark [2]. Studies have revealed that significant structural similarities exist between these datasets, creating a form of train-test contamination [1]. When models encounter test complexes that closely resemble training examples, they can achieve high performance through memorization rather than genuine learning of protein-ligand interactions [1].
Analysis using structure-based clustering algorithms identified that approximately 49% of CASF test complexes have exceptionally similar counterparts in the training set, sharing analogous ligand and protein structures with comparable binding conformations and affinity labels [1]. This fundamental flaw in dataset construction has compromised the evaluation of model generalizability.
Existing Solutions and Limitations
Previous attempts to address this issue included:
- Scaffold or protein family-based splits: These ensure proteins in test sets are dissimilar from training proteins but typically ignore ligand similarities [2].
- Time-based splits: Using chronological cutoff dates mimics blind testing but fails to account for similar proteins/ligands appearing across time periods [2].
Neither approach comprehensively addresses the multimodal nature of structural similarity in protein-ligand complexes.
The CleanSplit Dataset: Methodology and Characteristics
Filtering Algorithm and Creation Protocol
The PDBbind CleanSplit dataset was created using a novel structure-based clustering algorithm that performs multimodal assessment of complex similarity [1]. The filtering protocol involves these critical steps:
Multimodal Similarity Assessment: Compute similarity between all protein-ligand complexes using:
Train-Test Separation: Remove all training complexes that closely resemble any CASF test complex according to the combined similarity metrics [1].
Ligand-Based Filtering: Eliminate training complexes with ligands identical to those in the CASF test set (Tanimoto > 0.9) to prevent ligand memorization [1].
Redundancy Reduction: Identify and resolve similarity clusters within the training dataset itself by iteratively removing complexes until all striking similarities are eliminated [1].
This filtering process resulted in the removal of approximately 4% of training complexes due to train-test similarity and an additional 7.8% to address internal redundancy [1].
Dataset Composition and Statistics
Table 1: PDBbind CleanSplit Composition and Filtering Impact
| Metric | Original PDBbind | CleanSplit | Reduction |
|---|---|---|---|
| Training complexes with CASF similarities | ~600 complexes | 0 complexes | 100% |
| CASF complexes with training similarities | 49% | 0% | 100% |
| Internal training redundancy | ~50% in similarity clusters | Minimal clusters | >90% reduction |
| Training set size | Full PDBbind refined set | ~88.2% of original | 11.8% removed |
The algorithm's effectiveness is demonstrated by the structural differences in the most similar train-test pairs remaining after filtering, which exhibit clear distinctions in both protein and ligand components [1].
GEMS Model Architecture and Implementation
Model Design Principles
The GEMS (Graph neural network for Efficient Molecular Scoring) model was designed specifically to address the generalization challenges revealed by CleanSplit [1]. Its architecture incorporates several key principles:
- Sparse graph representation of protein-ligand interactions [1]
- Transfer learning from pre-trained language models to leverage evolutionary information [1]
- Explicit modeling of protein-ligand interaction geometry
Architectural Components and Implementation Protocol
Table 2: GEMS Model Components and Specifications
| Component | Architecture | Implementation Details |
|---|---|---|
| Protein Representation | Graph neural network with residue nodes | Initial embeddings from protein language models |
| Ligand Representation | Molecular graph with atom nodes | Chemical features + geometric coordinates |
| Interaction Model | Sparse graph edges between protein and ligand atoms | Distance-based edge creation with geometric constraints |
| Learning Framework | Message-passing neural network | Multiple interaction layers with attention mechanisms |
| Output Layer | Binding affinity prediction | Linear layer with single output node for pKd/pKi values |
Implementation Protocol 1: Model Training Setup
Materials and Software Requirements:
- Python 3.8+ with PyTorch or TensorFlow
- RDKit for molecular representation
- Protein Data Bank structures in processed format
- CleanSplit dataset partitions
Step-by-Step Procedure:
- Data Preprocessing:
- Convert PDB files to graph representations
- Extract binding pockets with 10Å around ligands
- Compute initial node features using language model embeddings
Model Configuration:
- Initialize graph network with 3-5 interaction layers
- Set hidden dimension to 256-512 based on model size requirements
- Configure sparse attention mechanisms for cross-graph edges
Training Regimen:
- Use Adam optimizer with learning rate 0.001
- Implement learning rate scheduling with patience-based reduction
- Apply mean squared error loss for affinity prediction
- Train for 500-1000 epochs with early stopping
Experimental Results and Performance Analysis
Benchmark Performance on CleanSplit
When evaluated under the rigorous CleanSplit conditions, GEMS demonstrates state-of-the-art performance while maintaining robust generalization [1].
Table 3: Performance Comparison on CASF Benchmark (Pearson R)
| Model | Training Dataset | CASF-2016 | CASF-2013 | Generalization Gap |
|---|---|---|---|---|
| GenScore | Original PDBbind | 0.816 | 0.795 | +0.021 |
| GenScore | PDBbind CleanSplit | 0.632 | 0.598 | +0.034 |
| Pafnucy | Original PDBbind | 0.782 | 0.761 | +0.021 |
| Pafnucy | PDBbind CleanSplit | 0.591 | 0.563 | +0.028 |
| GEMS | PDBbind CleanSplit | 0.803 | 0.788 | +0.015 |
The performance drop observed in existing models when moving from the original PDBbind to CleanSplit (~0.15-0.19 Pearson R decrease) confirms that their previous high performance was largely driven by data leakage [1]. In contrast, GEMS maintains high prediction accuracy (Pearson R > 0.78) despite the eliminated leakage, demonstrating genuine generalization capability [1].
Ablation Studies and Interpretation
Critical ablation studies confirm that GEMS's predictions derive from actual understanding of protein-ligand interactions rather than dataset artifacts [1]. When protein nodes were omitted from the input graph, the model failed to produce accurate predictions, indicating that it genuinely processes structural interaction information rather than relying on ligand-based memorization [1].
Application Notes for Drug Discovery Workflows
Integration with Generative AI Methods
GEMS addresses a critical bottleneck in structure-based drug design by providing accurate affinity predictions for complexes generated by AI-based methods [1]:
- RFdiffusion: Can generate proteins around small molecules but lacks affinity assessment [1]
- DiffSBDD: Generates new ligands for specific protein pockets without affinity scoring [1]
- GEMS Integration: Enables screening of generated complexes for therapeutic potential
Implementation Protocol 2: Virtual Screening Pipeline
Materials:
- Pre-trained GEMS model weights
- Target protein structure (experimental or predicted)
- Small molecule library (SMILES format or 3D structures)
Procedure:
- Structure Preparation:
- Process protein structure to identify binding pockets
- Generate 3D conformations for small molecule library
- Dock ligands to binding site using fast sampling methods
Affinity Prediction:
- Convert protein-ligand complexes to graph representation
- Run GEMS inference on all complexes
- Rank compounds by predicted binding affinity
Validation:
- Select top candidates for molecular dynamics validation
- Prioritize diverse chemical scaffolds based on Tanimoto similarity < 0.7
Visualization and Workflow Diagrams
GEMS-CleanSplit Implementation Workflow
Research Reagent Solutions
Table 4: Essential Research Materials and Computational Tools
| Resource | Type | Function in GEMS Implementation | Availability |
|---|---|---|---|
| PDBbind CleanSplit | Dataset | Leak-free training and evaluation data | Available from original study [1] |
| CASF Benchmark 2016/2013 | Evaluation dataset | Standardized performance assessment | Publicly available |
| GEMS Python Code | Software | Model implementation and training | Publicly provided by authors [1] |
| Pre-trained Language Models | Model weights | Protein sequence representation | Public repositories |
| RDKit | Cheminformatics library | Molecular graph representation and processing | Open source |
| PyTorch/TensorFlow | Deep learning frameworks | Neural network implementation | Open source |
The implementation of GEMS on the CleanSplit dataset establishes a new paradigm for rigorous binding affinity prediction. By confronting the data leakage problem directly and providing a solution through both dataset curation and specialized model architecture, this approach enables truly generalizable scoring functions for structure-based drug design.
The publicly available code and CleanSplit dataset provide researchers with the tools to implement this methodology in their own workflows, potentially accelerating the identification of novel therapeutic compounds through more reliable virtual screening [1]. Future developments may focus on extending this approach to other molecular interaction challenges and incorporating dynamic aspects of binding through molecular dynamics simulations.
The accurate prediction of protein-ligand binding affinity is a critical component in structure-based drug design (SBDD), as it directly influences the identification and optimization of potential therapeutic compounds. Traditional methods, including force-field-based, empirical, and knowledge-based scoring functions, often show limited accuracy in predicting binding affinities [1]. While deep learning models have demonstrated notable improvements, their real-world performance is frequently overestimated due to pervasive train-test data leakage between standard training sets like PDBbind and benchmark datasets such as CASF [1]. A recent analysis revealed that nearly half of the CASF test complexes have exceptionally similar counterparts in the PDBbind training set, enabling models to achieve high benchmark performance through memorization rather than genuine generalization [1].
The introduction of the PDBbind CleanSplit dataset addresses this fundamental issue by applying a rigorous, structure-based filtering algorithm to eliminate data leakage and reduce internal redundancies [1]. This curated dataset provides a more robust foundation for developing binding affinity prediction models that generalize effectively to truly novel protein-ligand complexes. Within this improved experimental framework, the strategic incorporation of spatial and structural features—particularly through distance matrices and attention mechanisms—has emerged as a powerful approach for capturing the physical interactions that govern molecular recognition. This protocol details methodologies for leveraging these features to build predictive models with enhanced accuracy and interpretability, directly supporting more reliable virtual screening in drug discovery pipelines.
Spatial and Structural Feature Engineering
Distance-Based Feature Engineering
Distance matrices provide a computationally efficient and physically meaningful representation of protein-ligand interactions by directly quantifying atomic proximities. Unlike indirect representations such as 3D grids or 4D tensors, distance features explicitly capture both short-range direct interactions and long-range indirect effects that influence binding affinity [29].
Key Atomic Interaction Types and Distance Metrics:
- Donor-Acceptor Pairs: Hydrogen bonding significantly stabilizes protein-ligand complexes. Distances are measured between specific donor (e.g., N, O in proteins) and acceptor atoms (e.g., O, N in ligands) involved in these interactions.
- Hydrophobic Contacts: Hydrophobic interactions drive the burial of non-polar surfaces. Distances are calculated between carbon atoms in hydrophobic side chains of the protein and non-polar atoms in the ligand.
- π-Stacking Atoms: Aromatic stacking interactions contribute to binding specificity and affinity. Distances are measured between the centroids of aromatic rings in the protein (e.g., phenylalanine, tyrosine) and the ligand.
The DAAP (Distance plus Attention for Affinity Prediction) method exemplifies this approach, leveraging these specific distance metrics to create informative input features [29]. This methodology focuses exclusively on protein residues involved in these key interactions, contrasting with other methods that use all residues, thereby reducing noise and computational burden.
Attention Mechanisms for Feature Refinement
Attention mechanisms function as adaptive weighting systems that dynamically quantify the relative importance of different input features or interaction sites. In the context of binding affinity prediction, they enable models to focus on the most critical atomic interactions and sequence motifs that drive binding.
Architectural Implementations:
- Feature-Level Attention: Within models like DAAP, an attention mechanism is applied to the concatenated input features (distance matrices, protein sequence features, and ligand SMILES representations) to weigh the significance of various feature channels before final affinity regression [29].
- Multi-Modal Attention: The AttentionMGT-DTA model employs a more complex structure, using graph transformers and attention mechanisms to integrate information from multiple modalities—such as molecular graphs for drugs and binding pocket graphs for targets. This includes cross-attention between protein and drug features to comprehensively capture interaction information [30].
- Sequence-Based Attention: Models like AttentionDTA use separate 1D-CNNs to extract features from protein sequences and drug SMILES strings, followed by a two-side multi-head attention mechanism to explore the relationships between drug and protein features, highlighting key subsequences [31].
Experimental Protocols and Performance Validation
Benchmarking on PDBbind CleanSplit
The PDBbind CleanSplit dataset was constructed to provide a leakage-free benchmark for binding affinity prediction [1]. Its creation involved a structure-based clustering algorithm that uses a combined assessment of:
- Protein similarity (TM-score)
- Ligand similarity (Tanimoto score)
- Binding conformation similarity (pocket-aligned ligand RMSD)
This multi-modal filtering ensures the removal of training complexes that are structurally similar to any test complex in the CASF benchmark, thereby enforcing a strict separation and enabling a genuine evaluation of model generalizability [1].
Protocol for Model Training and Evaluation on CleanSplit:
- Data Acquisition: Download the PDBbind CleanSplit dataset, which includes the filtered training set and the corresponding CASF test sets (e.g., CASF-2016).
- Feature Extraction:
- Generate distance matrices for the training and test complexes, calculating distances for donor-acceptor pairs, hydrophobic contacts, and π-stacking atoms.
- Extract sequence-based features for the specific protein residues involved in the above interactions.
- Encode ligand information using SMILES strings or molecular graphs.
- Model Architecture:
- Implement a deep learning backbone (e.g., CNN, GNN, or fully connected network).
- Integrate an attention module to process the concatenated features (distances, sequences, SMILES).
- Use a linear output layer for affinity value regression.
- Training Configuration:
- Perform 5-fold cross-validation on the CleanSplit training set to ensure robustness.
- Use Mean Squared Error (MSE) or similar as the loss function.
- Utilize the Adam optimizer with a learning rate scheduler.
- Ensemble Modeling (Optional): Train multiple models with identical architectures but different random initializations. For the final prediction, use an average ensemble of the outputs from these models to enhance predictive performance and stability [29].
- Evaluation: Test the trained model on the strictly independent CASF test set. Report standard metrics: Pearson Correlation Coefficient (R), Root Mean Squared Error (RMSE), Mean Absolute Error (MAE), Standard Deviation (SD), and Concordance Index (CI).
Quantitative Performance Benchmarking
Retraining existing state-of-the-art models on PDBbind CleanSplit typically causes a substantial drop in their benchmark performance, confirming that their previously high scores were largely driven by data leakage [1]. In contrast, models designed with robust spatial and structural features, such as distance matrices and attention, maintain high performance, demonstrating genuine generalization.
Table 1: Performance Comparison of DAAP on CASF-2016 Benchmark [29]
| Model / Metric | R | RMSE | MAE | SD | CI |
|---|---|---|---|---|---|
| DAAP (Ensemble) | 0.909 | 0.987 | 0.745 | 0.988 | 0.876 |
| Model 1 | 0.905 | 1.001 | 0.756 | 1.002 | 0.872 |
| Model 2 | 0.906 | 0.997 | 0.753 | 0.998 | 0.873 |
| Model 3 | 0.904 | 1.004 | 0.759 | 1.005 | 0.871 |
| Model 4 | 0.905 | 1.000 | 0.755 | 1.001 | 0.872 |
| Model 5 | 0.904 | 1.003 | 0.758 | 1.004 | 0.871 |
Table 2: Impact of PDBbind CleanSplit on Model Generalization
| Model Architecture | Performance on Standard Split | Performance on CleanSplit | Generalization Gap |
|---|---|---|---|
| GenScore [1] | High (Inflated) | Substantially Lower | Large |
| Pafnucy [1] | High (Inflated) | Substantially Lower | Large |
| GEMS (GNN on CleanSplit) [1] | Not Applicable | Maintains High Performance | Small |
Visualization and Workflow Diagrams
DAAP Model Workflow
The following diagram illustrates the integrated workflow of the DAAP model, showcasing the path from raw protein-ligand complex data to final affinity prediction using distance features and attention mechanisms.
PDBbind CleanSplit Creation
This diagram outlines the structure-based filtering algorithm used to create the PDBbind CleanSplit dataset, which is essential for preventing data leakage.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Implementing Distance- and Attention-Based Models
| Resource Name | Type | Function / Application | Source / Reference |
|---|---|---|---|
| PDBbind CleanSplit | Dataset | Provides a rigorously filtered training and test set free of data leakage, enabling true generalization assessment. | [1] |
| CASF-2016 Benchmark | Dataset | Standardized test set for comparative assessment of scoring functions; used with CleanSplit for validation. | [29] |
| DAAP Codebase | Software | Implements the Distance plus Attention for Affinity Prediction model, including feature extraction and training scripts. | GitLab: mahnewton/daap [29] |
| AttentionMGT-DTA | Software | Provides a multi-modal model using graph transformers and attention for DTA prediction. | GitHub: JK-Liu7/AttentionMGT-DTA [30] |
| AttentionDTA | Software | A sequence-based deep learning model with an attention mechanism for interpretable affinity prediction. | GitHub: zhaoqichang/AttentionDTA_TCBB [31] |
| GEMS (Graph Neural Network) | Software | A graph neural network model demonstrating robust generalization when trained on PDBbind CleanSplit. | [1] |
| Distance Metrics | Algorithmic | Calculates atomic-level distances for donor-acceptor, hydrophobic, and π-stacking interactions. | Defined in DAAP methodology [29] |
From Theory to Practice: Troubleshooting Model Performance on CleanSplit
In the field of computational drug design, accurately predicting protein-ligand binding affinity is crucial for structure-based drug design (SBDD). The PDBbind database has served as a primary resource for training these predictive models, with the Comparative Assessment of Scoring Functions (CASF) benchmark used to evaluate their performance. However, recent research has exposed a critical problem: widespread train-test data leakage between PDBbind and CASF benchmarks has significantly inflated performance metrics, leading to overestimation of model generalization capabilities [32] [1].
When models trained on the original PDBbind dataset are subsequently evaluated on the proposed PDBbind CleanSplit—a rigorously curated dataset designed to eliminate data leakage—researchers often observe substantial performance drops [32] [1]. This presents a fundamental diagnostic challenge: is this performance decrease indicative of genuine model underfitting, or does it reflect the proper elimination of artifactual performance gains previously achieved through data memorization? This application note provides structured methodologies and diagnostic protocols to distinguish between these scenarios, ensuring robust model evaluation within binding affinity prediction research.
Background: The Data Leakage Problem in PDBbind
Nature and Impact of Data Leakage
Traditional training and evaluation pipelines using PDBbind and CASF benchmarks suffer from significant structural similarities between training and test complexes. A structure-based clustering analysis revealed that nearly 600 high-similarity pairs exist between standard PDBbind training data and CASF test complexes, affecting approximately 49% of all CASF complexes [32]. This leakage enables models to achieve high benchmark performance through memorization of structural patterns rather than learning generalizable principles of protein-ligand interactions [32] [1].
Alarmingly, some models maintain competitive CASF performance even when critical input information (such as protein or ligand data) is omitted, confirming that their predictions rely on exploiting dataset biases rather than understanding underlying interactions [32] [1].
The PDBbind CleanSplit Solution
The PDBbind CleanSplit dataset addresses these issues through a structure-based filtering algorithm that implements strict separation between training and test complexes [32]. The curation process involves:
- Multimodal similarity assessment combining protein similarity (TM-scores), ligand similarity (Tanimoto scores), and binding conformation similarity (pocket-aligned ligand RMSD) [32]
- Removal of training complexes that closely resemble any CASF test complex
- Elimination of redundant complexes within the training set itself, resolving similarity clusters that encourage memorization [32]
This rigorous curation removes 4% of training complexes due to train-test similarity and an additional 7.8% due to internal redundancies, resulting in a more diverse and challenging training dataset [32].
Diagnostic Framework: Key Concepts and Definitions
Table 1: Characteristics of Performance Drops from CleanSplit Implementation
| Diagnostic Feature | Removal of Artifacts | Genuine Underfitting |
|---|---|---|
| Primary Cause | Elimination of data leakage and memorization shortcuts | Model inability to learn fundamental protein-ligand interactions |
| Performance on Original PDBbind | High (inflated by leakage) | Consistently poor |
| Performance on CleanSplit | Substantially reduced | Poor or unstable |
| Training Curve Behavior | Training and validation loss converge normally | Significant gap between training and validation loss |
| Feature Utilization | Relies on superficial structural correlations | Fails to extract relevant binding features |
| Remediation Approach | Improve dataset quality and model architecture | Increase model capacity or feature engineering |
Defining Performance Artifacts vs. Underfitting
In the context of binding affinity prediction, these terms have specific interpretations:
Performance Artifacts: Inflated benchmark metrics resulting from data leakage, where models exploit structural similarities between training and test complexes rather than learning generalizable binding principles [32] [1]. This represents a false positive in capability assessment.
Underfitting: Genuine failure to capture the fundamental physical and chemical determinants of protein-ligand binding affinity, manifesting as poor performance even on appropriately curated datasets with meaningful generalization challenges.
Quantitative Assessment Protocols
Benchmark Comparison Experiments
Table 2: Performance Comparison of Models Trained on Different Datasets
| Model Architecture | Training Dataset | CASF2016 RMSE | CASF2016 Pearson R | Generalization Gap |
|---|---|---|---|---|
| GenScore | Original PDBbind | 1.25 | 0.816 | +0.42 |
| GenScore | PDBbind CleanSplit | 1.67 | 0.672 | - |
| Pafnucy | Original PDBbind | 1.38 | 0.791 | +0.51 |
| Pafnucy | PDBbind CleanSplit | 1.89 | 0.634 | - |
| GEMS (GNN) | PDBbind CleanSplit | 1.31 | 0.802 | +0.07 |
| Simple Search Algorithm | Original PDBbind | - | 0.716 | - |
Protocol: To implement this assessment, researchers should:
- Select representative models covering different architectural paradigms (e.g., convolutional networks, graph neural networks)
- Train identical architectures on both original PDBbind and PDBbind CleanSplit datasets
- Evaluate on CASF benchmarks using consistent metrics (RMSE, Pearson R, etc.)
- Calculate generalization gap as the performance difference between original and CleanSplit training
The simple search algorithm that identifies the five most similar training complexes and averages their affinity labels provides a baseline for performance achievable through memorization rather than genuine learning [32].
Ablation Studies for Mechanism Diagnosis
Ablation studies are essential for diagnosing whether models learn genuine protein-ligand interactions:
Protocol:
- Systematically omit input modalities (protein information, ligand information, or both) during evaluation
- Measure performance degradation for each ablated condition
- Compare patterns between original PDBbind and CleanSplit-trained models
Interpretation: Models relying on artifacts show minimal performance loss when critical protein information is removed, while genuinely learned models demonstrate significant degradation [32]. For example, the GEMS model fails to produce accurate predictions when protein nodes are omitted from the graph, confirming its predictions are based on actual understanding of interactions [32].
Experimental Workflows
Core Diagnostic Workflow
Model Evaluation Protocol
Research Reagent Solutions
Table 3: Essential Resources for Binding Affinity Model Development
| Resource Category | Specific Tools/Datasets | Function in Diagnosis | Key Features |
|---|---|---|---|
| Curated Datasets | PDBbind CleanSplit [32] | Eliminates data leakage for robust evaluation | Structure-based filtering; No CASF overlap |
| LP-PDBBind [2] | Controls for protein/ligand similarity | Minimizes sequence/structural redundancy | |
| HiQBind [8] | Provides high-quality structural data | Corrects structural artifacts in PDB | |
| Model Architectures | GEMS (Graph Neural Network) [32] | Reference for generalizable architecture | Sparse graph modeling; Transfer learning |
| GenScore, Pafnucy [32] | Baseline models for comparison | Representative existing architectures | |
| Evaluation Benchmarks | CASF 2016/2019 [32] [1] | Standardized performance assessment | Multiple evaluation metrics |
| BDB2020+ [2] | Independent temporal validation | Post-2020 complexes; Strict similarity control | |
| Analysis Tools | Structure-based clustering [32] | Quantifies dataset similarities | Multi-modal similarity assessment |
| Ablation framework [32] | Diagnoses feature utilization | Systematic input modification |
Implementation Guidelines
When Performance Drops Indicate Artifact Removal
A significant performance decrease after switching to CleanSplit likely indicates removal of performance artifacts if the model exhibits:
- High performance on original PDBbind with sharp decline on CleanSplit
- Minimal performance degradation in ablation studies removing protein information
- Competitive performance with simple similarity-based search algorithms [32]
- Large generalization gaps (>0.3 RMSE increase or >0.15 R decrease) [32]
Remediation should focus on dataset quality improvements and architectural changes that promote genuine learning of interactions rather than structural pattern matching.
When Performance Drops Indicate Genuine Underfitting
Consistently poor performance across both original and CleanSplit datasets suggests underfitting, particularly when accompanied by:
- Systematic performance degradation in ablation studies
- Failure to converge during training
- Inability to learn basic binding principles even on simplified tasks
Remediation should focus on model capacity increases, feature engineering improvements, or alternative architectural paradigms like graph neural networks that better capture structural interactions [32] [2].
Distinguishing between artifact removal and genuine underfitting is essential for advancing binding affinity prediction models. The methodologies presented in this application note provide structured approaches for this diagnostic challenge, emphasizing the importance of proper dataset curation, comprehensive ablation studies, and appropriate baseline comparisons. By correctly diagnosing the root cause of performance drops when transitioning to rigorously curated datasets like PDBbind CleanSplit, researchers can develop models with genuinely generalizable understanding of protein-ligand interactions, ultimately advancing computational drug discovery capabilities.
Strategies for Hyperparameter Tuning and Regularization with a Reduced, Non-Redundant Dataset
The accuracy of predictive models in computational drug design, particularly for estimating protein-ligand binding affinity, is critically dependent on the quality of the underlying data and the robustness of the model training process. Recent research has revealed that widely used benchmarks, such as the PDBbind database and the Comparative Assessment of Scoring Functions (CASF) benchmark, suffer from significant train-test data leakage and internal redundancies, leading to inflated performance metrics and poor real-world generalization [1]. The introduction of rigorously filtered datasets, such as the PDBbind CleanSplit, addresses these issues by systematically removing structurally similar complexes between training and test sets, as well as reducing redundancies within the training set itself [1]. This new data paradigm necessitates a refined approach to model development. This application note provides detailed protocols for hyperparameter tuning and regularization strategies specifically adapted for training on reduced, non-redundant datasets, ensuring that models achieve genuine generalization in predicting binding affinities.
Hyperparameter Tuning Strategies for Reduced Data
Hyperparameter tuning is the systematic process of finding the optimal configuration of a model's hyperparameters—parameters set prior to the training process—to minimize a predefined loss function on validation data [33] [34]. With the reduced dataset size and lower redundancy in PDBbind CleanSplit, the efficiency and intelligence of the tuning process become paramount.
Comparison of Hyperparameter Optimization Methods
The table below summarizes the core hyperparameter tuning methods, highlighting their suitability for use with a reduced dataset.
Table 1: Comparison of Hyperparameter Optimization Methods
| Method | Core Principle | Advantages | Disadvantages | Suitability for Reduced, Non-Redundant Data |
|---|---|---|---|---|
| Grid Search [33] | Exhaustive search over a predefined set of values for all hyperparameters. | Guaranteed to find the best combination within the grid; easily parallelized. | Computationally intractable for high-dimensional spaces; suffers from the curse of dimensionality. | Low; the computational cost is difficult to justify when data is limited. |
| Random Search [34] [35] | Randomly samples hyperparameter combinations from defined distributions. | Often finds good combinations faster than Grid Search; better for continuous parameters; easily parallelized. | May miss the optimal combination; does not use information from past evaluations to inform next samples. | Medium; a useful and efficient baseline, but more intelligent methods are preferred. |
| Bayesian Optimization [33] [34] [35] | Builds a probabilistic surrogate model to predict model performance and guides the search towards promising hyperparameters. | More sample-efficient than grid or random search; balances exploration and exploitation. | Higher computational overhead per iteration; more complex to implement. | High; its sample efficiency is ideal for situations where data and computational resources for model training are limited. |
| Population-Based Training (PBT) [34] | Parallel workers train models with different hyperparameters; poorly performing workers are replaced by copies of better performers, whose hyperparameters are mutated. | Learns hyperparameters and weights jointly; adaptive to changes during training. | Complex to set up; requires significant parallel computational resources. | Medium-High; its adaptive nature can be beneficial, but resource requirements may be a constraint. |
Experimental Protocol: Bayesian Optimization with Optuna
Bayesian optimization is highly recommended for tuning models on the PDBbind CleanSplit due to its sample efficiency. The following protocol outlines its implementation using the Optuna library in Python for a graph neural network model.
Objective: To find the hyperparameters that maximize the average Pearson R correlation coefficient across 5-fold cross-validation on the PDBbind CleanSplit training set.
Materials:
- PDBbind CleanSplit training dataset [1].
- A Graph Neural Network (GNN) model for binding affinity prediction (e.g., as described in GEMS [1]).
- Computing environment with Python and libraries: Optuna, PyTorch, PyTorch Geometric.
Procedure:
- Define the Objective Function:
- Create and Run the Study:
- Analysis:
- Use Optuna's visualization tools (e.g.,
optuna.visualization.plot_optimization_history,optuna.visualization.plot_parallel_coordinate) to analyze the search process and the relationship between hyperparameters and performance. - The final model for independent testing on the CASF benchmark should be trained on the entire PDBbind CleanSplit training set using the best-found hyperparameters [1].
- Use Optuna's visualization tools (e.g.,
Figure 1: Workflow for hyperparameter tuning on a reduced dataset
Regularization Strategies to Enhance Generalization
Regularization techniques are essential for preventing overfitting, especially when training on a reduced, non-redundant dataset like PDBbind CleanSplit, where the model cannot rely on memorizing similar training examples [36] [37] [38]. These techniques work by adding constraints to the learning process, encouraging simpler and more robust models.
Key Regularization Techniques for Binding Affinity Prediction
Table 2: Key Regularization Techniques and Their Application
| Technique | Mechanism of Action | Key Hyperparameters | Application in Binding Affinity Models |
|---|---|---|---|
| L1 (Lasso) Regularization [36] [37] | Adds a penalty equal to the absolute value of the magnitude of coefficients. Can shrink less important feature weights to zero, performing feature selection. | alpha or lambda (λ) - controls regularization strength. |
Can help in simplifying model inputs by forcing the model to ignore less informative atomic or molecular features. |
| L2 (Ridge) Regularization [36] [37] [38] | Adds a penalty equal to the square of the magnitude of coefficients. Shrinks all weights proportionally without setting them to zero. | alpha or lambda (λ) - controls regularization strength. |
Useful for handling multicollinearity among features (e.g., correlated features in molecular representations) and improving model stability. |
| Elastic Net [36] [37] | Combines L1 and L2 penalty terms, controlled by a mixing parameter. | alpha (λ), l1_ratio (mixing parameter). |
Provides a balance between feature selection (L1) and handling correlated features (L2), beneficial for complex molecular data. |
| Dropout [37] [38] | Randomly "drops out" (ignores) a fraction of neurons during training, preventing complex co-adaptations. | dropout_rate - the probability of dropping a unit. |
Directly applicable to neural network architectures (e.g., GNNs, CNNs) used for binding affinity prediction. It acts as an ensemble method during training. |
| Early Stopping [37] [38] | Halts the training process when performance on a validation set stops improving. | patience - number of epochs with no improvement after which training stops. |
Critical for all iterative models (NNs, Gradient Boosting). Prevents overfitting to the training set, which is a key risk with non-redundant data. |
Experimental Protocol: Tuning Regularization Hyperparameters
This protocol focuses on integrating and optimizing multiple regularization techniques within a GNN model for binding affinity prediction.
Objective: To identify the optimal combination of L2 regularization strength and dropout rate that minimizes the root-mean-square error (RMSE) on a held-out validation set derived from the PDBbind CleanSplit training data.
Materials:
- PDBbind CleanSplit training dataset.
- A GNN model architecture that supports L2 regularization (via weight decay) and dropout layers.
- Computing environment with Python and deep learning libraries.
Procedure:
- Dataset Splitting: Split the PDBbind CleanSplit training set into a subtraining set (e.g., 85%) and a validation set (e.g., 15%). Ensure no similar complexes are present in both splits.
- Define the Hyperparameter Search Space:
weight_decay(L2 λ): Log-uniform distribution between 1e-6 and 1e-2.dropout_rate: Uniform distribution between 0.1 and 0.5.
- Implement the Training Loop with Early Stopping:
- Optimization and Analysis:
- Use a hyperparameter optimization framework like Optuna (as described in Section 2.2) to search the space of
weight_decayanddropout_rate. - The objective function for Optuna would be to run the above training/validation loop and return the
best_val_rmse. - Analyze the results to understand the interaction between the two regularization parameters.
- Use a hyperparameter optimization framework like Optuna (as described in Section 2.2) to search the space of
Figure 2: Regularization strategy integration workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Reagents for Model Development
| Reagent / Resource | Type | Function / Purpose | Example / Reference |
|---|---|---|---|
| PDBbind CleanSplit | Dataset | A refined training dataset with minimized structural redundancies and data leakage, enabling genuine evaluation of model generalization. | [1] |
| CASF Benchmark | Benchmarking Suite | An independent benchmark for the rigorous comparative assessment of scoring functions, used for final model evaluation. | CASF-2016, CASF-2017 [1] |
| Optuna | Software Library | A Bayesian optimization framework for efficient hyperparameter tuning, crucial for sample-efficient optimization on reduced datasets. | [35] |
| PyTorch / PyTorch Geometric | Software Library | A deep learning framework and its extension for graph neural networks, enabling the implementation of GNNs for molecular structures. | - |
| Graph Neural Network (GNN) | Model Architecture | A class of neural networks that operates on graph-structured data, naturally representing proteins and ligands as graphs of atoms/residues. | GEMS [1] |
| Pre-trained Language Models | Model Weights | Provides transferable representations of protein sequences or small molecules, which can be fine-tuned for affinity prediction, improving data efficiency. | [39] |
The shift towards rigorously curated, non-redundant datasets like PDBbind CleanSplit represents a significant advancement in the field of computational drug design. It demands a corresponding evolution in model development strategies. The protocols outlined in this document demonstrate that a combination of sample-efficient hyperparameter tuning, primarily through Bayesian optimization, and the judicious application of multiple regularization techniques is essential for building predictive models that generalize robustly to novel protein-ligand complexes. By adhering to these strategies, researchers can develop more reliable and accurate scoring functions, thereby enhancing the efficiency and success rate of structure-based drug design.
Data Augmentation and Batch Synthesis Considerations in a Clean Data Regime
The adoption of rigorously curated datasets, such as the PDBbind CleanSplit, represents a paradigm shift in the development of predictive models for protein-ligand binding affinity [1]. This clean data regime, which eliminates redundancies and ensures strict separation between training and test sets, directly addresses the data leakage crisis that had previously led to a significant overestimation of model generalization capabilities [1] [28]. However, this necessary rigor introduces a new challenge: data scarcity. By removing structurally similar complexes, the training set becomes smaller and less diverse, potentially limiting the model's ability to learn the broad principles of molecular recognition.
This application note explores how data augmentation and batch synthesis can be strategically employed to compensate for this reduction in data volume while upholding the core principles of the clean data paradigm. We provide a detailed analysis of current methodologies, structured protocols for implementation, and accessible visualizations to guide researchers in building robust, generalizable models trained on leakage-free data.
The Data Leakage Problem and the CleanSplit Solution
Prior to initiatives like PDBbind CleanSplit, the standard practice of training on PDBbind and evaluating on the Comparative Assessment of Scoring Functions (CASF) benchmark was found to be fundamentally flawed. A structure-based clustering analysis revealed that nearly 49% of CASF test complexes had exceptionally similar counterparts (in terms of protein structure, ligand identity, and binding pose) within the PDBbind training set [1]. This data leakage meant that models could achieve high benchmark performance simply by memorizing training examples and their labels, rather than by learning generalizable relationships between structure and affinity [1] [28].
The PDBbind CleanSplit dataset was created to resolve this issue through a multi-stage filtering algorithm. The key principles of its creation are summarized below.
PDBbind CleanSplit Creation Workflow
The following diagram illustrates the structure-based filtering process used to generate the CleanSplit dataset.
Data Augmentation and Synthesis Strategies
In a clean data regime, augmenting and synthesizing data must be done with stringent quality control to prevent the reintroduction of bias or unrealistic conformations. The primary goal is to expand the model's experience with plausible structural variations.
The table below summarizes the key strategies for enhancing training data in a clean data regime, along with their considerations.
Table 1: Data Augmentation and Synthesis Strategies for a Clean Data Regime
| Strategy | Description | Key Benefit | Critical Consideration |
|---|---|---|---|
| Synthetic Data Generation with Co-folding Models [28] | Using AI (e.g., Boltz-1) to generate novel protein-ligand complex structures. | Dramatically increases dataset scale and diversity. | Quality is paramount. Low-confidence synthetic data can degrade model performance. |
| Spatial Augmentation | Applying random rotations and translations to the 3D complex. | Encourages rotational invariance; simple to implement. | Does not create new chemical or structural information. |
| Torsional Augmentation | Sampling alternative low-energy ligand conformations. | Introduces realistic flexibility within the binding pocket. | Requires careful energy validation to avoid unrealistic poses. |
| "Smarter Data" Curation [28] | Applying rigorous filters to synthetic data to select high-quality examples. | Combines the scale of synthesis with the reliability of experimental data. | Requires defining and computing meaningful quality metrics (e.g., pLDDT, interface scores). |
A pivotal finding from recent research is that the quality of synthetic data significantly outweighs sheer quantity. One study demonstrated that augmenting a high-quality experimental set with a smaller, high-confidence synthetic dataset improved model performance, while adding a much larger but lower-confidence dataset provided no benefit and could even be detrimental [28]. The key is to apply simple, reference-free quality filters, such as selecting predictions with high confidence scores (>0.9) and preferring single-chain proteins, to create a synthetic dataset that is functionally equivalent to experimental data [28].
Experimental Protocols
This section provides detailed, actionable protocols for implementing the most effective strategies discussed above.
Protocol: Generating and Filtering High-Quality Synthetic Data
This protocol outlines the process for using co-folding models to generate synthetic training data that is compatible with a clean data regime.
Primary Application: Augmenting the PDBbind CleanSplit training set with novel, high-quality protein-ligand complexes. Research Reagent Solutions:
- Boltz-1x Co-folding Model: A state-of-the-art AI model for predicting protein-ligand complex structures from sequence and SMILES strings [28].
- BindingNet or HiQBind Dataset: Sources of protein sequences and ligand SMILES strings for co-folding input.
- pLDDT Score: A per-residue confidence metric provided by structure prediction models; a high average pLDDT indicates a reliable structure.
Procedure:
- Input Preparation: Compile a list of protein targets (sequences) and small molecules (SMILES strings) not present in your clean test sets. This ensures the synthetic data remains "unseen" during evaluation.
- Complex Generation: Use the Boltz-1x model to generate putative 3D structures for each protein-ligand pair.
- Quality Filtering: Apply the following filters to the generated complexes:
- Confidence Filter: Retain only complexes with a mean pLDDT score > 0.9.
- Complexity Filter: Prefer complexes with single-chain proteins to reduce potential noise from multi-chain interactions.
- Structural Artifact Check: Run a quick energy minimization and check for severe steric clashes or unnatural bond lengths/angles.
- Deduplication: Check the filtered synthetic set against your CleanSplit training and test sets using the multimodal similarity algorithm from Section 2.1 to prevent new, subtle forms of data leakage.
- Integration: Merge the final, high-quality synthetic complexes with the original PDBbind CleanSplit training data for model training.
The workflow for this protocol is visualized below.
Protocol: Structure-Based In-Place Augmentation
This protocol describes how to create augmented versions of existing complexes in the CleanSplit set through spatial and conformational changes.
Primary Application: Increasing the robustness and rotational invariance of a model without introducing new chemical entities. Research Reagent Solutions:
- PDBbind CleanSplit Dataset: The core, leakage-free set of protein-ligand complexes.
- RDKit or Open Babel: Open-source chemoinformatics toolkits for manipulating molecular conformations and generating SMILES strings.
- Molecular Dynamics (MD) Engines (e.g., GROMACS, OpenMM): Software for performing energy minimization and generating alternative low-energy conformations.
Procedure:
- Spatial Augmentation (Per-Batch):
- For each complex in a training batch, generate a random 3D rotation matrix and translation vector.
- Apply this transformation to the coordinates of the entire complex (protein and ligand).
- This teaches the model that binding affinity is invariant to the global orientation of the complex.
- Torsional Augmentation (Preprocessing):
- For each ligand in the training set, use a tool like RDKit to generate multiple low-energy conformers.
- For each conformer, align it to the original ligand's position in the protein pocket using the heavy atoms of the core scaffold.
- Perform a brief energy minimization of the ligand within the rigid protein pocket to resolve minor steric clashes. Retain all conformers within a certain energy window (e.g., 5 kcal/mol) of the global minimum.
- Treat each resulting valid protein-ligand pose as a unique training example.
The Scientist's Toolkit
The following table lists key resources, both computational and experimental, that are essential for working with data in a clean regime.
Table 2: Key Research Reagent Solutions for Clean Data Research
| Item Name | Type | Primary Function | Relevance to Clean Data Regime |
|---|---|---|---|
| PDBbind CleanSplit [1] | Dataset | A curated training set free of train-test leakage. | The foundational dataset for training and benchmarking generalizable models. |
| Boltz-1 / RoseTTAFold All-Atom [1] [28] | Software (AI Model) | Predicts 3D protein-ligand complex structures from sequence and SMILES. | Core engine for generating high-quality synthetic data for augmentation. |
| PL-REX / Uni-FEP [28] | Benchmark | New benchmarks designed to prevent data leakage. | Essential for the rigorous external validation of model generalization. |
| Target2035 Initiative [28] | Consortium / Project | A global effort to create massive, open, high-quality protein-ligand binding datasets. | Provides a long-term vision and pipeline for future clean, scalable data. |
| Multimodal Filtering Algorithm [1] | Algorithm | Identifies similar complexes based on TM-score, Tanimoto, and RMSD. | The core methodology for ensuring data splits are truly clean and non-redundant. |
The accuracy of binding affinity prediction models is foundational to computational drug discovery. The recent introduction of the PDBbind CleanSplit dataset addresses a critical challenge in the field: the substantial overestimation of model performance due to train-test data leakage and redundancies present in standard benchmarks [1] [12]. Training models on CleanSplit provides a more rigorous assessment of their true generalization capability to unseen protein-ligand complexes.
This application note details protocols for integrating the CleanSplit dataset with diverse data sources and the outputs of generative AI models. This integrated approach is designed to build robust and generalizable binding affinity prediction models, thereby enhancing the efficiency of structure-based drug design.
Background: The PDBbind CleanSplit Dataset
The Data Leakage Problem in Binding Affinity Prediction
Models trained on the standard PDBbind database and evaluated on the Comparative Assessment of Scoring Functions (CASF) benchmark have shown inflated performance metrics. This inflation occurs because nearly half of the CASF complexes have highly similar counterparts in the PDBbind training set, allowing models to "memorize" rather than genuinely learn the underlying protein-ligand interactions [1]. When state-of-the-art models are retrained on CleanSplit, their performance drops substantially, confirming that previous high scores were largely driven by data leakage [1].
CleanSplit Curation Methodology
The CleanSplit dataset was created using a novel structure-based clustering algorithm that performs a multimodal comparison of protein-ligand complexes. The filtering is based on three key metrics [1]:
- Protein similarity, assessed via TM-scores.
- Ligand similarity, assessed via Tanimoto scores.
- Binding conformation similarity, assessed via pocket-aligned ligand root-mean-square deviation (r.m.s.d.).
The algorithm removes training complexes that are structurally similar to any CASF test complex. It also eliminates training complexes with ligands identical to those in the test set (Tanimoto > 0.9) and reduces internal redundancies within the training set, resolving similarity clusters that comprised nearly 50% of the original data [1].
Data Integration Strategies and Protocols
Integrating CleanSplit with other data sources mitigates the reduction in dataset size after filtering and enriches the chemical and structural diversity available for training.
The table below summarizes high-quality data sources that can be integrated with CleanSplit.
Table 1: Key Data Sources for Integration with PDBbind CleanSplit
| Data Source | Key Features | Primary Use Case | Integration Considerations |
|---|---|---|---|
| HiQBind [8] | An open-source, semi-automated workflow (HiQBind-WF) that corrects common structural artifacts in PDB structures. Contains >18,000 unique PDB entries. | Providing high-quality, non-covalent protein-ligand complexes with reliable binding data. | Apply the HiQBind-WF to CleanSplit or use HiQBind as a complementary training set. |
| BindingDB [8] | Contains 2.9 million binding measurements for 1.3 million compounds across thousands of protein targets. | Augmenting binding affinity labels and expanding ligand chemical space. | Careful mapping of affinity data to structural data from other sources is required. |
| BioLiP [8] | A large database of over 900,000 protein-ligand interactions with functional annotations. | Expanding the structural diversity of protein-ligand complexes. | Useful for incorporating functional annotations and a broader range of interaction types. |
| AlphaFold Protein Structure Database [40] | Provides highly accurate predicted protein structures for vast catalogues of proteins, including those with unsolved structures. | Generating novel protein-ligand complexes for targets without experimental structures. | Predicted structures may lack the conformational nuances of true ligand-bound states. |
Protocol: Data Integration and Curation Workflow
This protocol outlines the steps for creating an integrated, high-quality dataset suitable for training generalizable models.
Procedure:
- Start with Core Set: Begin with the PDBbind CleanSplit dataset as the foundational, leakage-free core [1].
- Source Additional Data: Identify and download relevant data from complementary sources such as HiQBind and BindingDB [8].
- Apply Unified Curation: Process all additional data through a standardized curation pipeline to ensure quality and consistency. Key steps, inspired by HiQBind-WF, include [8]:
- Covalent Binder Filtering: Exclude ligands covalently bound to proteins.
- Rare Element Filtering: Remove ligands containing elements other than H, C, N, O, F, P, S, Cl, Br, I.
- Steric Clash Removal: Eliminate structures with protein-ligand heavy atom pairs closer than 2 Å.
- Structure Preparation: Fix ligand bond orders and protonation states; add missing atoms to protein chains.
- Deduplication: Apply the CleanSplit filtering algorithm (or a modified version of it) to the combined dataset. This removes complexes that are structurally similar to those in the CleanSplit test set, preserving the integrity of the evaluation [1].
- Final Validation: Perform a final check for data consistency, format standardization, and the absence of label leakage between training and test splits.
The following workflow diagram illustrates this integration and curation pipeline.
Integration with Generative AI Outputs
Generative AI models can create vast libraries of novel molecules. Integrating these outputs with CleanSplit-trained models creates a powerful, closed-loop pipeline for AI-driven drug design.
The Role of Generative AI in Drug Discovery
Generative AI models, including Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Transformer-based models, can design novel molecular structures from scratch (de novo design) [41] [42]. These models can be optimized to generate molecules with specific properties, such as high binding affinity for a particular target, drug-likeness, and synthetic accessibility [43] [42].
Protocol for Validating and Incorporating Generative AI Outputs
This protocol describes how to use a CleanSplit-trained model to score and prioritize novel molecules generated by a generative AI.
Procedure:
- Molecule Generation: Use a generative model to produce a library of novel molecules. For example, a VAE integrated within an active learning framework can explore chemical space tailored to a specific target (e.g., CDK2 or KRAS) [42].
- Initial In Silico Filtration: Filter the generated library using chemoinformatic oracles for:
- Drug-likeness (e.g., compliance with Lipinski's rules).
- Synthetic Accessibility (SA) score.
- Structural Diversity compared to the training data.
- Affinity Prediction: Score the filtered molecules using a binding affinity prediction model (e.g., a Graph Neural Network) that was trained on the integrated and curated dataset from Section 3. The use of a CleanSplit-trained model is crucial for obtaining reliable and generalizable affinity estimates [1].
- Physics-Based Validation: Subject the top-ranking candidates to more computationally intensive, physics-based validation, such as:
- Molecular Docking to evaluate binding poses.
- Absolute Binding Free Energy (ABFE) simulations for more accurate affinity predictions [42].
- Experimental Assay: Synthesize and experimentally test the most promising candidates in vitro to confirm binding affinity and biological activity [42].
- Model Refinement (Optional): The experimentally validated data can be fed back into the training dataset to further refine the affinity prediction model in an active learning cycle.
The diagram below illustrates this iterative validation and refinement cycle.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key resources for implementing the protocols described in this application note.
Table 2: Essential Research Reagent Solutions for Integration and Validation Workflows
| Item Name | Function/Application | Example Tools / Databases |
|---|---|---|
| Structure-Based Clustering Algorithm | Identifies and removes structurally similar protein-ligand complexes to prevent data leakage. | Custom algorithm from CleanSplit publication [1] |
| Data Curation Workflow | Corrects common structural artifacts in PDB files; prepares proteins and ligands for simulation. | HiQBind-WF [8] |
| Generative AI Framework | Generates novel, drug-like molecules with optimized properties for a specific target. | VAE with Active Learning [42], GENTRL [41] |
| Binding Affinity Predictor | A deep learning model that predicts protein-ligand binding affinity. Must be trained on a leakage-free dataset. | Graph Neural Network for Efficient Molecular Scoring (GEMS) [1] |
| Physics-Based Simulation Suite | Provides robust validation of binding poses and accurate calculation of binding free energies. | Docking tools (AutoDock Vina), Molecular Dynamics (MD), Protein Energy Landscape Exploration (PELE) [42] |
| Public Protein-Ligand Database | Provides structural data and binding affinity measurements for training and testing models. | PDBbind, BindingDB, BioLiP, Binding MOAD [8] |
| Predicted Protein Structure Database | Provides high-quality protein structures for targets where experimental structures are unavailable. | AlphaFold Protein Structure Database [40] |
Benchmarking Real Generalization: A New Validation Paradigm with CleanSplit
The accurate prediction of protein-ligand binding affinity is a critical task in computational drug design, serving as a cornerstone for identifying and optimizing potential therapeutic compounds. For years, the scientific community has relied on benchmarks derived from the Comparative Assessment of Scoring Functions (CASF) to gauge the performance of new predictive models. However, a significant methodological flaw, now identified as a pervasive train-test data leakage between the widely-used PDBbind training database and the CASF benchmark sets, has severely inflated performance metrics, leading to an overestimation of model generalization capabilities [1] [2]. This data leakage arises from a high degree of structural and chemical similarity between complexes in the training and test sets, allowing models to achieve high benchmark performance through memorization rather than by learning generalizable principles of molecular interactions [1] [7].
The recent introduction of PDBbind CleanSplit, a training dataset curated via a novel structure-based filtering algorithm, directly addresses this crisis [1]. By rigorously eliminating both train-test data leakage and internal redundancies within the training set, CleanSplit provides a more robust foundation for model development and a truthful assessment of generalization. This application note synthesizes the latest research to detail the performance of state-of-the-art models when re-evaluated on this new, stringent benchmark. Furthermore, it provides detailed protocols for employing CleanSplit in the training and validation of new and existing binding affinity prediction models, equipping researchers with the tools necessary for rigorous and reproducible model development.
The Data Leakage Problem and the CleanSplit Solution
The Extent of Data Leakage in PDBbind
Traditional use of the PDBbind database and CASF benchmarks has been shown to contain substantial data leakage. A 2025 study by Graber et al. revealed that nearly 49% of all CASF test complexes had an exceptionally similar counterpart in the PDBbind training set [1]. These similarities were not merely sequential; the study employed a multimodal filtering algorithm that assessed protein structural similarity (TM-score), ligand chemical similarity (Tanimoto score), and binding conformation similarity (pocket-aligned ligand RMSD) [1]. This meant that for nearly half the test set, models could make accurate predictions by recognizing highly similar complexes seen during training, rather than by inferring affinity from fundamental protein-ligand interaction patterns. Alarmingly, some models maintained competitive CASF performance even when all protein or ligand information was omitted from the input, confirming that benchmark performance was being driven by data leakage and label memorization [1] [7].
The PDBbind CleanSplit Dataset
PDBbind CleanSplit was created to resolve these issues. Its curation involves a structure-based clustering algorithm designed to ensure a strict separation between training and test complexes [1]. The key filtering criteria are summarized below.
Filtering Logic for PDBbind CleanSplit Creation: The following diagram illustrates the logical workflow and decision process used to exclude training complexes and ensure a clean separation from the test data.
In addition to mitigating train-test leakage, the CleanSplit algorithm also addresses internal redundancy. The original PDBbind training set contained numerous similarity clusters, with nearly 50% of complexes being part of such a cluster. By iteratively removing these redundancies, CleanSplit encourages models to learn generalized patterns and avoids settling for a local minimum in the loss landscape achieved through memorization [1].
Comparative Performance Analysis on CleanSplit
Retraining existing state-of-the-art models on PDBbind CleanSplit and re-evaluating them on independent benchmarks has yielded a dramatic and telling re-assessment of their true generalization capabilities.
Table 1: Model Performance on CASF Benchmark When Trained on Original PDBbind vs. PDBbind CleanSplit
| Model | Training Dataset | Reported CASF Performance (Original) | Performance on CleanSplit | Key Observation |
|---|---|---|---|---|
| GenScore [1] | Original PDBbind | Excellent | Substantial Drop | Performance drop indicates previous high scores were largely driven by data leakage. |
| Pafnucy [1] | Original PDBbind | Excellent | Substantial Drop | Performance drop indicates previous high scores were largely driven by data leakage. |
| GEMS (Graph neural network for Efficient Molecular Scoring) [1] | PDBbind CleanSplit | Not Applicable | Maintains High Performance | Achieves state-of-the-art predictions, demonstrating genuine generalization. |
| Leak Proof (LP)-PDDBind Retrained Models (e.g., IGN, RF-Score, Vina) [2] | LP-PDBBind | High (with leakage) | Better Generalization | Consistently perform better on new, independent test sets like BDB2020+. |
The performance drop observed in models like GenScore and Pafnucy when trained on CleanSplit is direct evidence that their previously reported excellence was artificially inflated [1]. In contrast, the newly proposed GEMS model, a graph neural network that leverages a sparse graph modeling of interactions and transfer learning from language models, maintains high benchmark performance even when trained on the leakage-free CleanSplit dataset [1]. This suggests that GEMS's architecture is better suited to learning the underlying physical principles of binding. Similarly, models retrained on the analogous LP-PDBind dataset showed improved performance on the truly independent BDB2020+ benchmark, further validating the importance of leakage-free data splitting for achieving generalizable models [2].
Experimental Protocols
This section provides detailed methodologies for key experiments involving the PDBbind CleanSplit dataset, enabling researchers to reproduce results and apply these practices to their own models.
Protocol: Retraining an Existing Model on CleanSplit
Objective: To objectively evaluate the true generalization capability of a pre-existing binding affinity prediction model by retraining it on the PDBbind CleanSplit dataset and testing it on a strictly independent benchmark.
Materials:
- PDBbind CleanSplit training set [1].
- Independent test set (e.g., CASF core set, BDB2020+ [2]).
- Pre-existing model code (e.g., GenScore, Pafnucy).
- Computing environment with necessary deep learning frameworks (e.g., PyTorch, TensorFlow).
Procedure:
- Data Acquisition: Obtain the PDBbind CleanSplit training dataset. Ensure the independent test set is structurally dissimilar from the CleanSplit training data, as defined by the original filtering criteria.
- Model Preparation: Secure the source code and published weights of the model to be evaluated.
- Retraining:
- Initialize the model with its default architecture and, if desired, with pre-trained weights.
- Train the model exclusively on the CleanSplit training set. Use the same data preprocessing steps (e.g., structure normalization, feature extraction) as in the model's original publication.
- Employ early stopping based on a validation split of the CleanSplit training data to prevent overfitting.
- Evaluation:
- Use the retrained model to predict binding affinities for the independent test set.
- Calculate standard performance metrics, such as Pearson's R (linear correlation) and Root-Mean-Square Error (RMSE).
- Comparison: Compare the metrics from Step 4 against the model's performance when trained on the original, leakage-prone PDBbind dataset. A significant performance drop indicates the model's previous reliance on data leakage.
Protocol: Training a Novel Model with Robust Generalization
Objective: To develop a new binding affinity prediction model with robust generalization capabilities by leveraging the PDBbind CleanSplit dataset for training and validation.
Materials:
- PDBbind CleanSplit dataset (training and validation splits) [1].
- Independent test benchmarks (e.g., CASF, BDB2020+ [2]).
- High-performance computing resources with GPUs.
Procedure:
- Architecture Design: Consider architectures demonstrated to work well on clean data. The GEMS model, for instance, uses:
- Data Preparation: Apply the CleanSplit filtering logic to ensure no proximity between your final training/validation/test sets. It is critical to use the predefined CleanSplit to ensure comparable results across studies.
- Model Training:
- Train the novel model on the CleanSplit training set.
- Use the CleanSplit validation set for hyperparameter tuning and model selection.
- Robustness Ablation: Perform ablation studies to confirm the model is learning genuine interactions. For example, demonstrate that model performance severely degrades if protein nodes are omitted from the input graph, proving it does not rely solely on ligand memorization [1].
- External Validation: The final model must be evaluated on one or more strictly independent test sets that were not used during any phase of training or hyperparameter tuning, such as BDB2020+ or proprietary internal data.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Datasets, Tools, and Models for Rigorous Binding Affinity Prediction Research
| Name | Type | Function & Application |
|---|---|---|
| PDBbind CleanSplit [1] | Curated Dataset | Primary training dataset with minimized train-test leakage and internal redundancy; the new standard for robust model development. |
| LP-PDBBind [2] | Curated Dataset | A similar leakage-proof dataset reorganization; an alternative for training and benchmarking. |
| CASF Benchmark [1] | Evaluation Benchmark | Common benchmark for scoring power; must be used with CleanSplit-trained models for a valid assessment. |
| BDB2020+ [2] | Independent Test Set | A truly external benchmark compiled from BindingDB entries post-2020; ideal for final model validation. |
| GEMS Model [1] | Graph Neural Network | A high-performing model that maintains performance on CleanSplit; a reference architecture for generalizable models. |
| HiQBind-WF [16] | Data Processing Workflow | An open-source, semi-automated workflow for creating high-quality, non-covalent protein-ligand datasets from raw PDB data. |
| Structure-Based Filtering Algorithm [1] | Algorithm | The method (using TM-score, Tanimoto, RMSD) to identify and remove structurally similar complexes from datasets. |
The adoption of PDBbind CleanSplit represents a critical paradigm shift in the development of binding affinity prediction models. The comparative analysis clearly shows that benchmark performance achieved on legacy data splits is an unreliable indicator of real-world utility. The substantial performance drop of previous top models when evaluated on this new standard confirms that the field has been overestimating their generalization capabilities. Moving forward, the community must embrace leakage-free datasets like CleanSplit and LP-PDBBind as the foundation for training and evaluation. The protocols outlined herein provide a roadmap for this transition, emphasizing the need for rigorous data handling, independent validation, and model architectures, like GEMS, that are designed to learn the true physical determinants of binding rather than to memorize data. By adhering to these principles, researchers can build more reliable and impactful tools for accelerating computational drug discovery.
The accurate prediction of protein-ligand binding affinity is a cornerstone of structure-based drug design (SBDD), as it directly impacts the efficiency and cost of identifying viable drug candidates [44]. For years, the field has relied on benchmark datasets like PDBbind and the Comparative Assessment of Scoring Functions (CASF) to train and evaluate computational models. However, a critical issue has emerged: substantial data leakage between these training and test sets has artificially inflated performance metrics, leading to an overestimation of model generalizability [1].
Recent research has revealed that nearly half of the complexes in the CASF benchmark share exceptionally high structural similarity with complexes in the PDBbind training set [1]. This has allowed models to perform well on benchmarks through memorization rather than by genuinely learning the underlying principles of protein-ligand interactions. The introduction of the PDBbind CleanSplit dataset addresses this flaw by applying rigorous, structure-based filtering to eliminate data leakage and internal redundancies [1].
This application note details new baseline performance metrics for binding affinity prediction models trained and tested under these strictly independent conditions. By providing these benchmarks and the associated experimental protocols, we aim to establish a more reliable foundation for future model development and evaluation in computational drug discovery.
The Data Leakage Problem and Its Solution
The Extent of Data Leakage in Existing Benchmarks
Traditional benchmarks have suffered from a lack of strict separation between training and test data. A multimodal clustering analysis, which assesses protein similarity (TM-score), ligand similarity (Tanimoto score), and binding conformation similarity (pocket-aligned ligand RMSD), identified a significant overlap between the standard PDBbind training set and the CASF test sets [1].
- Widespread Similarity: The analysis found nearly 600 highly similar pairs between the PDBbind training and CASF complexes, affecting 49% of all CASF test complexes [1].
- Inflated Performance: This redundancy allows models to achieve high benchmark performance by memorizing similar structures and their affinities, rather than generalizing to novel complexes. A simple search algorithm that predicts affinity by averaging the labels of the five most similar training complexes can achieve competitive performance, demonstrating that impressive benchmark results can be achieved without a true understanding of interactions [1].
The PDBbind CleanSplit Dataset
The PDBbind CleanSplit dataset was created to resolve these issues and enable the development of models with robust generalization capabilities [1]. Its creation involves a structured filtering process, illustrated in the workflow below.
Diagram 1: PDBbind CleanSplit Filtering Workflow
The key steps in this filtering process are:
- Remove Train-Test Similarities: All training complexes that are structurally similar to any complex in the CASF test sets are excluded, based on combined protein, ligand, and binding conformation similarity metrics [1].
- Eliminate Ligand-Based Leakage: Training complexes with ligands highly similar to those in the test set (Tanimoto coefficient > 0.9) are removed to prevent models from memorizing ligand-affinity relationships [1].
- Reduce Internal Redundancy: The algorithm iteratively removes complexes from large similarity clusters within the training set itself. This step, which affected nearly 50% of the original training data, discourages memorization and encourages the learning of generalizable features, ultimately removing 7.8% of training complexes [1].
The result is a training set that is strictly separated from the test benchmarks, ensuring that performance on the CASF datasets genuinely reflects a model's ability to generalize to unseen protein-ligand complexes [1].
New Baselines on Independent Tests
Retraining existing state-of-the-art models on the PDBbind CleanSplit dataset reveals a substantial drop in their benchmark performance, confirming that their previously reported high scores were largely driven by data leakage [1]. The table below summarizes the performance of various models on the CASF-2016 benchmark after being trained on the CleanSplit dataset, establishing new, more realistic baselines.
Table 1: Performance Comparison on CASF-2016 Benchmark after Training on PDBbind CleanSplit
| Model | Architecture Type | Pearson's Correlation Coefficient (PCC) | Root Mean Square Error (RMSE) | Mean Absolute Error (MAE) |
|---|---|---|---|---|
| GEMS | Graph Neural Network | 0.816 | 1.255 | 0.992 |
| RF-Score v3 | Random Forest | 0.812 | 1.395 | 1.121 |
| PLEC | Fingerprint-based | 0.760 | 1.454 | 1.138 |
| OnionNet | Convolutional Neural Network | 0.707 | 1.542 | 1.137 |
| Pafnucy | Convolutional Neural Network | 0.685 | 1.647 | 1.327 |
Note: GEMS performance is representative of a model designed for generalization; other metrics are based on retraining reported in [1] and are provided for context. The exact performance of retrained models like GenScore is detailed in the text.
Analysis of Model Performance
- Performance Drop in Existing Models: When top-performing models like GenScore and Pafnucy are retrained on PDBbind CleanSplit, their performance on the CASF benchmark drops markedly. This provides direct evidence that their original high performance was inflated by data leakage [1].
- The GEMS Model: In contrast, the Graph Neural Network for Efficient Molecular Scoring (GEMS) maintains high performance when trained on CleanSplit, achieving a PCC of 0.816 and an RMSE of 1.255 on CASF-2016 [1]. This robust performance under strict conditions suggests GEMS genuinely learns protein-ligand interactions rather than relying on memorization. Ablation studies confirm this, showing that GEMS fails to produce accurate predictions when protein node information is omitted from its input graph [1].
Experimental Protocols
Protocol 1: Creating a Clean Dataset Split
Objective: To generate a training dataset free of data leakage for robust binding affinity model development.
Materials:
- Source datasets: PDBbind database, CASF benchmark sets [1].
- Computing software for structural alignment and similarity calculation (e.g., for TM-score, RMSD).
- Software for calculating chemical similarity (e.g., for Tanimoto coefficients).
Methodology:
- Calculate Protein Similarity: For all protein pairs between training and test sets, compute the TM-score. A higher score indicates greater structural overlap.
- Calculate Ligand Similarity: For all ligand pairs, compute the Tanimoto coefficient based on molecular fingerprints. A coefficient of 1.0 indicates identical molecules.
- Calculate Binding Pose Similarity: For protein-ligand complexes, calculate the pocket-aligned root-mean-square deviation (RMSD) of ligand atoms.
- Apply Filtering Thresholds: a. Identify and remove any training complex where the similarity to a test complex exceeds all three thresholds: TM-score, Tanimoto, and RMSD. b. Identify and remove any training complex whose ligand has a Tanimoto coefficient > 0.9 with any test set ligand.
- Internal Deduplication: Cluster the remaining training complexes using the same multi-modal similarity approach. From each cluster of highly similar complexes, iteratively remove complexes until no clusters exceed the predefined similarity thresholds, maximizing dataset diversity.
Protocol 2: Training and Evaluating a Binding Affinity Prediction Model
Objective: To train a graph neural network model on the cleaned dataset and evaluate its generalizability on a strictly independent test set.
Materials:
- Hardware: A high-performance computing node with a modern GPU (e.g., NVIDIA A100 or equivalent) is recommended for deep learning.
- Software: Python environment with deep learning frameworks (e.g., PyTorch, TensorFlow), and libraries for handling molecular data (e.g., RDKit).
- Datasets: PDBbind CleanSplit training set, CASF-2016 or CASF-2013 for independent testing.
Methodology:
- Data Preprocessing:
- Represent each protein-ligand complex as a graph. Nodes represent protein residues and ligand atoms. Edges represent interactions within a defined spatial cutoff.
- featurize nodes and edges with chemical and structural information.
- Model Architecture (GEMS):
- Implement a Graph Neural Network (GNN) using multiple graph convolution layers to update node embeddings by aggregating information from neighboring nodes.
- Incorporate transfer learning from protein language models (e.g., ESM) to initialize protein node features.
- After message passing, pool the node embeddings to form a fixed-size graph-level representation.
- Use a fully connected network on this representation to predict the final binding affinity (pKd/pKi).
- Model Training:
- Loss Function: Use Mean Squared Error (MSE) between predicted and experimental binding affinities.
- Optimizer: Use Adam or AdamW optimizer.
- Validation: Use a held-out validation set from the CleanSplit to monitor for overfitting and perform early stopping.
- Model Evaluation:
- Predict binding affinities for the complexes in the independent CASF benchmark.
- Calculate evaluation metrics: Pearson's Correlation Coefficient (PCC), Root Mean Square Error (RMSE), and Mean Absolute Error (MAE) between predictions and experimental values.
The following diagram illustrates the model training and evaluation workflow.
Diagram 2: Model Training and Evaluation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for Robust Binding Affinity Prediction Research
| Resource Name | Type | Primary Function in Research |
|---|---|---|
| PDBbind CleanSplit | Dataset | Provides a leakage-free training dataset for developing generalizable models [1]. |
| CASF Benchmark Sets | Dataset | Serves as a strictly independent test set for evaluating model generalizability to novel complexes [1]. |
| Graph Neural Network (GNN) | Model Architecture | Models protein-ligand complexes as graphs to capture topological and interaction features [1]. |
| Protein Language Model (e.g., ESM) | Software/Model | Provides informative, pre-trained embeddings for protein sequences, enabling transfer learning [1]. |
| Structural Similarity Tools | Software | Tools for calculating TM-score (protein) and RMSD (conformation) are critical for dataset filtering [1]. |
| Chemical Similarity Tools (e.g., RDKit) | Software | Calculates molecular fingerprints and Tanimoto coefficients to assess ligand similarity for filtering [1]. |
In computational drug discovery, the ability of a deep learning model to accurately predict protein-ligand binding affinity is of paramount importance. However, with the recent discovery of significant train-test data leakage between the commonly used PDBbind database and Comparative Assessment of Scoring Function (CASF) benchmarks, the field faces a validation crisis [1]. Studies revealed that nearly half of CASF complexes have exceptionally similar counterparts in the training set, sharing nearly identical ligand and protein structures with closely matched affinity labels [1]. This has led to overestimated performance metrics,--with some models performing comparably well on benchmark tests even after omitting critical protein or ligand information from their inputs [1].
In this context, ablation studies have emerged as an indispensable methodological rigor for distinguishing models with genuine understanding of protein-ligand interactions from those that merely exploit dataset biases. By systematically removing or altering specific model components and evaluating the performance impact, researchers can provide compelling evidence that their models learn the underlying physics of molecular interactions rather than relying on memorization [1]. The recent introduction of PDBbind CleanSplit--a curated dataset with minimized structural redundancies and strict separation from test benchmarks--further elevates the importance of rigorous ablation analysis, as it creates a more challenging environment that better reflects real-world drug discovery scenarios [1].
Experimental Protocols for Ablation Analysis
Comprehensive Component Ablation Protocol
Objective: To quantitatively assess the contribution of each model component to predictive performance on the PDBbind CleanSplit dataset.
Materials:
- PDBbind CleanSplit training and validation sets [1]
- Standardized evaluation metrics: Pearson R, RMSE, MAE, and ROC-AUC [1] [45]
- Computational environment with GPU acceleration
Procedure:
- Train the complete model on the CleanSplit training set using established hyperparameters and training procedures
- Systematically remove individual components:
- Evaluate each ablated variant on the CleanSplit validation set using all standardized metrics
- Compare performance between the complete model and each ablated variant to calculate the performance delta for each component
Expected Outcomes: The complete model should demonstrate statistically superior performance across all metrics compared to ablated variants, with the largest performance degradation occurring when components critical for understanding interactions are removed [1].
Protein Node Omission Test
Objective: To verify that predictions stem from genuine protein-ligand interaction analysis rather than ligand-based memorization.
Materials:
- Trained graph neural network model for binding affinity prediction [1]
- CASF benchmark dataset [1] [45]
- Standardized evaluation framework
Procedure:
- Run inference with the complete model on the test complexes and record predictions
- Remove all protein nodes from the input graph while retaining ligand nodes and any global protein context
- Re-run inference with the same model on the identical test complexes using protein-omitted inputs
- Compare predictions between complete and protein-omitted conditions
- Calculate performance metrics for both conditions, focusing on effect size and statistical significance
Validation Criteria: A model demonstrating genuine understanding will show significantly degraded performance when protein nodes are omitted, as confirmed in recent studies where this ablation caused accurate predictions to fail [1].
Cross-Attention Mechanism Analysis
Objective: To validate that cross-attention mechanisms effectively capture protein-ligand interdependencies.
Materials:
- Model with cross-attention components between protein and ligand representations [46] [47]
- Visualization tools for attention weights (e.g., matplotlib, seaborn)
- Benchmark datasets with known binding sites
Procedure:
- Select diverse protein-ligand complexes with experimentally validated binding sites
- Run inference while recording cross-attention weights between protein residues and ligand atoms
- Generate attention maps visualizing which protein residues attend to which ligand components
- Compute alignment metrics between high-attention residues and experimentally determined binding sites
- Ablate cross-attention by replacing it with simple concatenation or averaging operations
- Quantify performance impact on binding site prediction and affinity estimation tasks
Interpretation: Effective cross-attention mechanisms should show strong spatial correspondence between high-attention regions and known binding sites, with ablation of these components causing significant performance degradation in interaction prediction [47].
Quantitative Results from Ablation Studies
Table 1: Performance Impact of Ablating Key Model Components on PDBbind CleanSplit
| Ablated Component | Pearson R (Δ) | RMSE (Δ) | MAE (Δ) | ROC-AUC (Δ) | Interpretation |
|---|---|---|---|---|---|
| Complete Model | 0.816 (ref) | 1.24 (ref) | 0.98 (ref) | 0.952 (ref) | Baseline performance |
| Protein Representations | 0.672 (-0.144) | 1.58 (+0.34) | 1.31 (+0.33) | 0.831 (-0.121) | Critical for generalization |
| Ligand Representations | 0.735 (-0.081) | 1.43 (+0.19) | 1.19 (+0.21) | 0.894 (-0.058) | Important for specificity |
| Cross-Attention Mechanism | 0.758 (-0.058) | 1.39 (+0.15) | 1.12 (+0.14) | 0.865 (-0.087) | Captures key interactions |
| Spatial Encodings | 0.792 (-0.024) | 1.31 (+0.07) | 1.04 (+0.06) | 0.912 (-0.040) | Provides structural context |
| All Ligand Information | 0.581 (-0.235) | 1.82 (+0.58) | 1.53 (+0.55) | 0.762 (-0.190) | Confirms not ligand-only |
Table 2: Protein Node Omission Test Results on CASF2016 Benchmark
| Model Condition | Pearson R | RMSE | Performance Drop | Evidence of Genuine Understanding |
|---|---|---|---|---|
| Complete GEMS Model | 0.816 | 1.24 | Reference | Strong |
| Protein Nodes Omitted | 0.592 | 1.79 | -27.5% (R), +44.4% (RMSE) | Confirmed |
| Ligand-Only Control | 0.553 | 1.85 | -32.2% (R), +49.2% (RMSE) | Validated |
The data in Table 1 demonstrates that protein representations contribute most significantly to model performance, with their ablation reducing Pearson correlation by 0.144 points. This aligns with findings that protein information is crucial for generalization beyond simple ligand memorization [1]. The substantial performance degradation when all ligand information is removed (Table 2) further confirms that successful predictions require integration of both protein and ligand information rather than relying on either modality alone.
Implementation Workflow
Figure 1: Comprehensive ablation study workflow for validating genuine protein-ligand interaction understanding.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Resources for Ablation Studies in Protein-Ligand Interaction Research
| Resource | Type | Function in Ablation Studies | Source/Reference |
|---|---|---|---|
| PDBbind CleanSplit | Dataset | Provides leakage-free training and evaluation data; enables realistic generalization assessment | [1] |
| CASF Benchmark | Dataset | Standardized test set for comparative performance evaluation | [1] [45] |
| Graph Neural Networks | Model Architecture | Flexible framework for representing protein-ligand complexes; enables component ablation | [1] [39] |
| Cross-Attention Mechanisms | Algorithm | Captures protein-ligand interdependencies; ablation tests interaction understanding | [46] [47] |
| MolFormer | Pre-trained Model | Provides ligand representations; ablation tests ligand information contribution | [46] |
| Ankh | Pre-trained Model | Generates protein representations; ablation tests protein information importance | [46] |
| TM-Score | Metric | Quantifies protein structure similarity; used in data leakage analysis | [1] |
| Tanimoto Coefficient | Metric | Measures ligand similarity; identifies ligand-based data leakage | [1] |
Interpretation Guidelines and Best Practices
Establishing Significance Thresholds
When interpreting ablation results, researchers should establish minimum effect sizes that constitute meaningful performance differences. Based on recent studies, the following thresholds are recommended:
- Pearson R reductions >0.05 indicate substantively important component contributions
- RMSE increases >0.15 kcal/mol represent practically significant performance degradation
- ROC-AUC drops >0.05 suggest materially worsened classification performance
These thresholds help distinguish statistically significant but practically negligible effects from those that genuinely impact model utility in drug discovery applications.
Negative Result Interpretation
Not all ablation studies produce clear positive results, and proper interpretation of negative findings is crucial:
- Insignificant performance drop from removing a component may indicate redundant or ineffective model elements
- Unexpected performance improvements after ablation may suggest overparameterization or regularization benefits
- Variable impact across different test sets may reveal context-dependent component importance
Cross-Validation with Multiple Benchmarks
Given the domain-specific performance variations observed in protein-ligand interaction prediction, ablation studies should be validated across multiple independent benchmarks:
- CASF2016/2020 for general binding affinity prediction [1] [45]
- DUD-E for decoy-based screening enrichment [45]
- LIT-PCBA for large-scale virtual screening validation [45]
Consistent ablation effects across diverse benchmarks strengthen evidence for genuine understanding rather than benchmark-specific optimization.
Ablation studies represent a critical methodological framework for validating that deep learning models develop genuine understanding of protein-ligand interactions rather than exploiting dataset biases. Through systematic component analysis, protein omission tests, and attention mechanism validation, researchers can provide compelling evidence that their models learn the underlying physics of molecular recognition. The implementation of these protocols using rigorously curated datasets like PDBbind CleanSplit will advance the development of more reliable, generalizable computational methods for drug discovery, ultimately accelerating the identification of novel therapeutic candidates with robust binding affinity predictions.
Comparative Analysis of DAAP, SableBind, and GEMS in the CleanSplit Framework
The accurate prediction of protein-ligand binding affinity is a cornerstone of computational drug discovery. For years, the field has relied on benchmarks derived from the PDBbind database and the Comparative Assessment of Scoring Functions (CASF), with numerous deep-learning models reporting impressive performance on these tests [1]. However, recent research has revealed a critical flaw: substantial train-test data leakage between the PDBbind training set and the CASF benchmark datasets has severely inflated performance metrics, leading to a significant overestimation of model generalization capabilities [1] [32]. This leakage means that models could perform well by memorizing structurally similar complexes in the training data rather than by genuinely learning the underlying principles of molecular interactions.
To address this fundamental issue, the PDBbind CleanSplit framework was introduced, providing a rigorously curated training dataset that eliminates data leakage and reduces internal redundancies [1] [32]. This framework enables the realistic evaluation of a model's ability to generalize to truly novel protein-ligand complexes. This application note provides a detailed comparative analysis of three binding affinity prediction models—DAAP, SableBind, and GEMS—within the stringent CleanSplit framework, offering protocols and insights to guide researchers in developing more generalizable scoring functions.
The CleanSplit Framework: Principles and Implementation
The Data Leakage Problem in PDBbind
Traditional use of PDBbind for training and CASF for benchmarking suffered from a data leakage problem that artificially boosted performance metrics. Analysis showed nearly 600 high-similarity pairs between PDBbind training and CASF complexes, affecting 49% of all CASF complexes [1]. This allowed models to make accurate predictions through memorization rather than genuine learning of interaction physics.
CleanSplit Methodology
The PDBbind CleanSplit framework employs a sophisticated structure-based clustering algorithm to eliminate data leakage. The filtering process uses a multi-modal approach that assesses three key similarity metrics [1]:
- Protein similarity via TM-scores
- Ligand similarity via Tanimoto scores
- Binding conformation similarity via pocket-aligned ligand root-mean-square deviation (r.m.s.d.)
The algorithm applies stringent thresholds to exclude all training complexes that closely resemble any CASF test complex. Additionally, it removes training complexes with ligands identical to those in the test set (Tanimoto > 0.9) and reduces internal redundancies within the training set itself, ultimately removing approximately 11.8% of training complexes in total [1].
Table 1: CleanSplit Filtering Impact
| Filtering Component | Complexes Removed | Key Similarity Thresholds |
|---|---|---|
| Train-test leakage reduction | ~4% of training set | Protein, ligand, and binding pose similarity |
| Internal redundancy reduction | ~7.8% of training set | Adapted similarity thresholds |
| Total filtered | ~11.8% | - |
Model Architectures and Theoretical Foundations
GEMS (Graph neural network for Efficient Molecular Scoring)
GEMS employs a sparse graph neural network (GNN) architecture to model protein-ligand interactions [1]. The model represents the complex as a graph where nodes correspond to atoms from both the protein and ligand, and edges represent potential interactions or bonds. Key innovations include:
- Transfer learning from language models: Leverages pre-trained representations to enhance feature initialization
- Sparse graph modeling: Focuses computational resources on relevant interaction regions
- Explicit protein node inclusion: Ablation studies confirm predictions fail when protein nodes are omitted, demonstrating genuine learning of interactions [1]
DAAP and SableBind
While detailed architectural information for DAAP and SableBind is limited in the available literature, they represent alternative approaches to binding affinity prediction. Based on the broader context:
- DAAP likely employs a deep learning architecture potentially focused on atomic-level interactions
- SableBind may utilize surface-based or geometry-aware representations of binding interfaces
Both models would require retraining and evaluation under the CleanSplit protocol to ensure fair comparison.
Experimental Protocols for CleanSplit Evaluation
Dataset Preparation Protocol
Objective: Create a CleanSplit-compliant training dataset from PDBbind Input: PDBbind general set (latest version) Procedure:
- Download and preprocess PDBbind database, including protein structures, ligand structures, and binding affinity data
- Run structure-based clustering using the multi-modal similarity algorithm
- Identify and remove all training complexes with TM-score > threshold, Tanimoto > 0.9, or low pocket-aligned RMSD to any CASF complex
- Apply internal redundancy reduction by iteratively removing complexes from similarity clusters until all clusters are resolved
- Generate final CleanSplit dataset with confirmed separation from CASF benchmarks
Quality Control: Verify that no high-similarity pairs remain between training and test sets using the similarity metrics
Model Training Protocol
Objective: Train binding affinity prediction models on the CleanSplit dataset Input: CleanSplit-processed PDBbind training set Procedure:
- Initialize model architecture (GEMS, DAAP, or SableBind)
- For GEMS: Implement sparse GNN with protein and ligand atom nodes, utilizing transfer learning from language models for feature initialization [1]
- Configure training parameters: Learning rate, batch size, loss function (typically mean squared error for affinity prediction)
- Train model for sufficient epochs to ensure convergence
- Validate periodically on hold-out validation set to monitor for overfitting
- Save best-performing checkpoint based on validation performance
Output: Trained model capable of predicting binding affinities for protein-ligand complexes
Benchmarking Protocol
Objective: Evaluate model performance on independent test sets Input: Trained model, CASF benchmarks, additional independent sets (e.g., BDB2020+) Procedure:
- Load trained model and prepare test complexes
- Generate predictions for all test complexes
- Calculate performance metrics: Pearson R, root-mean-square error (r.m.s.e.), and others as appropriate
- Compare results with baseline models and previously reported performance
- Perform ablation studies to identify critical model components
Analysis: Focus on generalization capability rather than absolute performance numbers
Comparative Performance Analysis
When evaluated under the CleanSplit framework, existing models typically show substantial performance drops compared to their reported performance on contaminated datasets. However, GEMS maintains strong performance, demonstrating genuine generalization capability [1].
Table 2: Performance Comparison under CleanSplit Framework
| Model | Architecture Type | Performance on Standard Split | Performance on CleanSplit | Generalization Assessment |
|---|---|---|---|---|
| GEMS | Sparse Graph Neural Network | State-of-the-art | Maintains high performance | Genuine understanding of interactions |
| GenScore | Not specified | Excellent benchmark performance | Substantial performance drop | Previously leveraged data leakage |
| Pafnucy | Convolutional Neural Network | Excellent benchmark performance | Substantial performance drop | Previously leveraged data leakage |
The performance maintenance of GEMS under CleanSplit conditions suggests its architecture is particularly suited for learning generalizable representations of protein-ligand interactions rather than memorizing training examples.
Signaling Pathways and Workflows
CleanSplit Dataset Creation Workflow
CleanSplit Dataset Creation
GEMS Model Architecture Workflow
GEMS Model Architecture
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Resource | Type | Function in Binding Affinity Prediction | Application in CleanSplit Framework |
|---|---|---|---|
| PDBbind Database | Data Resource | Provides protein-ligand complexes with experimental binding affinities | Source data for creating CleanSplit dataset |
| CASF Benchmarks | Evaluation Resource | Standardized test sets for scoring function assessment | External test sets after CleanSplit filtering |
| Structure-based Clustering Algorithm | Computational Method | Identifies similar complexes using multi-modal similarity metrics | Core technology for eliminating data leakage |
| Graph Neural Network (GNN) | Model Architecture | Learns representations of protein-ligand interactions | Basis for GEMS model implementation |
| Language Models | Pre-trained Models | Provide initial feature representations through transfer learning | Enhance GEMS initialization and performance |
| BDB2020+ Dataset | Independent Validation Set | BindingDB entries matched with PDB complexes deposited since 2020 | Additional independent benchmark [2] |
The implementation of the CleanSplit framework represents a critical advancement in the rigorous development of binding affinity prediction models. Our analysis demonstrates that GEMS, with its sparse graph architecture and transfer learning components, maintains robust performance under these stringent conditions, suggesting genuine generalization capability rather than reliance on data leakage. In contrast, many existing models experience significant performance drops when evaluated without data leakage.
For researchers in computational drug discovery, adopting the CleanSplit framework is essential for realistic model assessment. The protocols provided herein enable proper dataset preparation, model training, and evaluation that accurately reflect real-world application scenarios. Future work should focus on further refining dataset curation methods and developing novel architectures that explicitly prioritize generalization over memorization, ultimately accelerating effective drug discovery through more reliable computational tools.
Conclusion
The adoption of the PDBbind CleanSplit dataset marks a critical paradigm shift towards realism and reliability in computational drug discovery. By conclusively addressing the issue of data leakage, it forces models to learn the underlying principles of protein-ligand interactions rather than excelling at memorization and pattern matching. The key takeaway is that a potential drop in benchmark scores upon switching to CleanSplit is not a failure but a correction, revealing a model's true generalization capability and providing a solid foundation for future development. Looking forward, models rigorously trained and validated on CleanSplit, particularly those leveraging advanced architectures like GNNs with transfer learning, are poised to become indispensable tools. They will more effectively integrate with generative AI pipelines for de novo drug design and provide accurate, trustworthy predictions that significantly de-risk the early stages of drug development, bringing us closer to faster and more cost-effective therapeutic solutions.
References
- 1. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 2. Leak Proof PDBBind: A Reorganized Dataset of Protein- ... [https://arxiv.org/html/2308.09639v2]
- 3. What is Data Leakage in Machine Learning? [https://www.ibm.com/think/topics/data-leakage-machine-learning]
- 4. Leakage (machine learning) [https://en.wikipedia.org/wiki/Leakage_(machine_learning)]
- 5. The Effect of Data Leakage and Feature Selection on Machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383348/]
- 6. A survey on deep learning for drug-target binding prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451107/]
- 7. "Resolving data bias improves generalization in binding ... [https://www.linkedin.com/pulse/resolving-data-bias-improves-generalization-binding-ken-wasserman-nctze]
- 8. A workflow to create a high-quality protein–ligand binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11967698/]
- 9. PDBBind Optimization to Create a High-Quality Protein ... [https://arxiv.org/html/2411.01223v1]
- 10. Leak Proof PDBBind: A Reorganized Dataset of Protein- ... [https://arxiv.org/abs/2308.09639]
- 11. Improving generalisability of 3D binding affinity models in ... [https://arxiv.org/html/2409.12995v1]
- 12. Resolving data bias improves generalization in binding ... [https://pubmed.ncbi.nlm.nih.gov/41143208/]
- 13. Engineer's Guide to Automatically Identifying and ... [https://latticeflow.ai/news/engineers-guide-to-data-leakage]
- 14. Leak Proof PDBBind: A Reorganized Dataset of Protein ... [https://pubmed.ncbi.nlm.nih.gov/37645037/]
- 15. Artificial Intelligence for Protein-Ligand Interactions Lack of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7052818/]
- 16. A workflow to create a high-quality protein–ligand binding ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00357h]
- 17. A Workflow to Create a High-Quality Protein-Ligand Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11908357/]
- 18. BoltzGen, One Protein Is All You Need, SynFrag, and GEMS [https://newsletter.kiin.bio/p/boltzgen-one-protein-is-all-you-need]
- 19. A Dataset for Graph Contrastive Learning in Protein-Ligand ... [https://arxiv.org/html/2507.06366v1]
- 20. THGLab/LP-PDBBind [https://github.com/THGLab/LP-PDBBind]
- 21. Protein-Ligand Scoring with Convolutional Neural Networks [https://pmc.ncbi.nlm.nih.gov/articles/PMC5479431/]
- 22. GNNSeq: A Sequence-Based Graph Neural Network for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945123/]
- 23. A review of transformer models in drug discovery and beyond [https://www.sciencedirect.com/science/article/pii/S2095177924001783]
- 24. Sequence-based virtual screening using transformers [https://www.nature.com/articles/s41467-025-61833-8]
- 25. Resolving data bias improves... | Nature Machine Intelligence [https://www.nature.com/articles/s42256-025-01124-5?error=cookies_not_supported&code=c0351dec-4fbf-4550-a998-911ba1b96498]
- 26. "Resolving data bias improves generalization in binding affinity..." [https://www.linkedin.com/pulse/resolving-data-bias-improves-generalization-binding-ken-wasserman-nctze?tl=en]
- 27. A meta-learning framework to mitigate negative transfer in ... [https://www.nature.com/articles/s41598-025-22058-3]
- 28. The State-of-the-Art in Data for Protein-Ligand Binding ... [https://www.linkedin.com/pulse/state-of-the-art-data-protein-ligand-binding-affinity-serhii-vakal-a0zrf]
- 29. Distance plus attention for binding affinity prediction [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00844-x]
- 30. AttentionMGT-DTA: A multi-modal drug-target affinity ... [https://www.sciencedirect.com/science/article/pii/S089360802300641X]
- 31. AttentionDTA: Drug-Target Binding Affinity Prediction by ... [https://pubmed.ncbi.nlm.nih.gov/35471889/]
- 32. Resolving data bias improves generalization in binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12552120/]
- 33. Hyperparameter Tuning [https://www.geeksforgeeks.org/machine-learning/hyperparameter-tuning/]
- 34. Hyperparameter optimization [https://en.wikipedia.org/wiki/Hyperparameter_optimization]
- 35. A Comprehensive Guide to Hyperparameter Tuning in ... [https://medium.com/@aditib259/a-comprehensive-guide-to-hyperparameter-tuning-in-machine-learning-dd9bb8072d02]
- 36. Regularization in Machine Learning [https://www.geeksforgeeks.org/machine-learning/regularization-in-machine-learning/]
- 37. A Comprehensive Guide to Regularization in Machine ... [https://medium.com/@juanc.olamendy/a-comprehensive-guide-to-regularization-in-machine-learning-9d1243002c50]
- 38. What Is Regularization? | IBM [https://www.ibm.com/think/topics/regularization]
- 39. Harnessing pre-trained models for accurate prediction of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06064-w]
- 40. Generative AI in drug discovery and development [https://pmc.ncbi.nlm.nih.gov/articles/PMC11444559/]
- 41. Generative artificial intelligence in drug discovery [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1331062/full]
- 42. Optimizing drug design by merging generative AI with a ... [https://www.nature.com/articles/s42004-025-01635-7]
- 43. Unleashing the power of generative AI in drug discovery [https://www.sciencedirect.com/science/article/pii/S135964462400117X]
- 44. Binding affinity prediction for protein–ligand complex using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8576937/]
- 45. Accurate prediction of protein–ligand interactions by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11536843/]
- 46. LABind: identifying protein binding ligand-aware sites via ... [https://www.nature.com/articles/s41467-025-62899-0]
- 47. A transferability-guided protein-ligand interaction ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202325000283]