Illumina vs. Nanopore Sequencing: A Strategic Comparison for Modern Chemogenomics and Drug Discovery
This article provides a comprehensive comparison of Illumina and Oxford Nanopore Technologies (ONT) sequencing platforms for chemogenomic applications.
Illumina vs. Nanopore Sequencing: A Strategic Comparison for Modern Chemogenomics and Drug Discovery
Abstract
This article provides a comprehensive comparison of Illumina and Oxford Nanopore Technologies (ONT) sequencing platforms for chemogenomic applications. Aimed at researchers and drug development professionals, it explores the foundational principles of each technology, their specific methodological applications in antimicrobial resistance and microbiome research, and practical guidance for troubleshooting and data optimization. By synthesizing recent comparative studies and validation metrics, this review serves as a strategic guide for selecting the appropriate sequencing platform to accelerate biomarker discovery, understand compound mechanisms of action, and advance personalized therapeutic development.
Core Sequencing Technologies: Principles, Evolution, and Relevance to Chemogenomics
{Article Content}
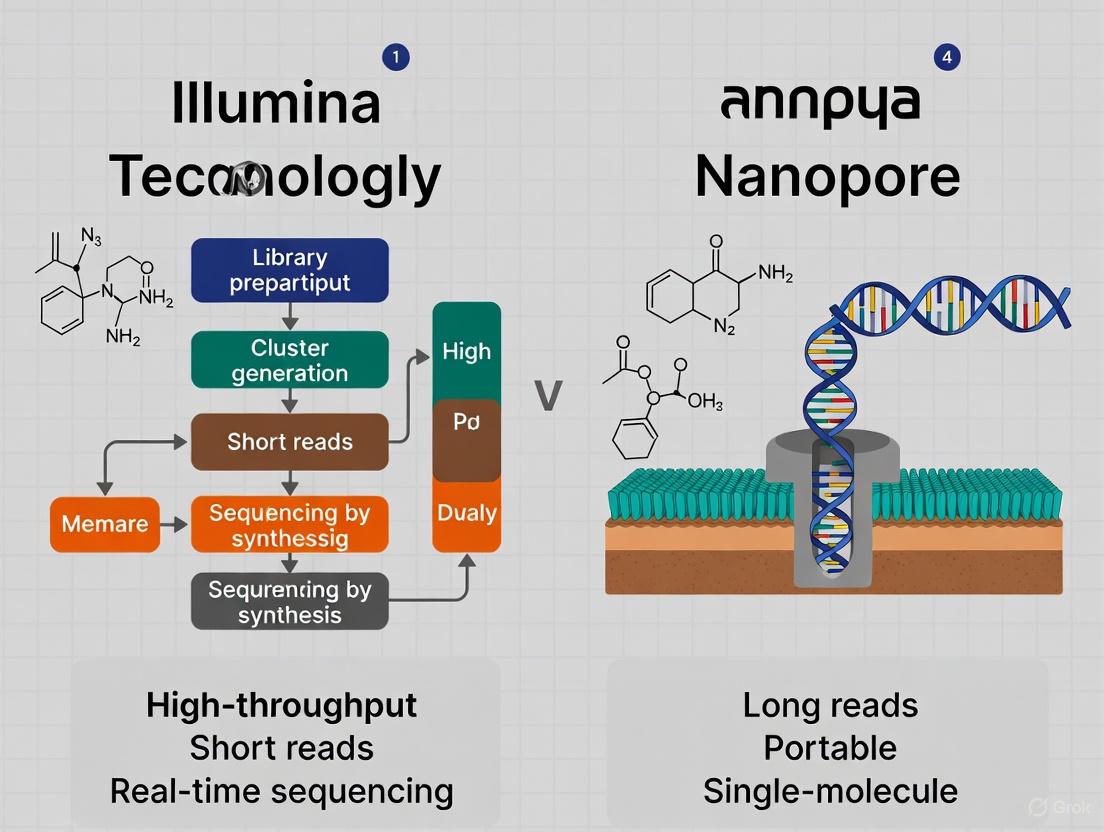
Illumina Short-Read Sequencing: High-Accuracy Reversible Terminator Chemistry
Next-generation sequencing (NGS) technologies are fundamental to modern chemogenomic research, enabling the exploration of how chemical compounds interact with biological systems. For applications ranging from target identification to understanding drug resistance mechanisms, the choice of sequencing platform directly impacts data quality and biological conclusions. Illumina's short-read sequencing, based on Sequencing by Synthesis (SBS) with reversible terminator chemistry, is renowned for its high accuracy and is often compared to Oxford Nanopore Technologies (ONT) long-read sequencing. This guide objectively compares the performance of these platforms, supported by experimental data, to inform researchers and drug development professionals in selecting the appropriate technology for their specific chemogenomic applications.
Technology Breakdown: Core Chemistries and Workflows
The fundamental difference between Illumina and Nanopore technologies lies in their underlying biochemistry and data acquisition methods, which in turn dictate their optimal application spheres.
Illumina Sequencing by Synthesis (SBS)
Illumina's SBS technology is a cyclic method that detects single bases as they are incorporated into growing DNA strands.
- Reversible Terminator Chemistry: Each of the four nucleotides (A, C, T, G) is labeled with a specific fluorescent dye and contains a reversible terminator that blocks the incorporation of the next nucleotide. This ensures a single base is added per cycle [1] [2].
- Imaging and Cleavage: After incorporation, the flow cell is imaged to determine the identity of the base based on its fluorescence. The terminator and fluorophore are then cleaved, allowing the incorporation of the next nucleotide [1]. This base-by-base sequencing virtually eliminates errors associated with homopolymer repeats (strings of identical bases) [2].
- Latest Advancements: The newest XLEAP-SBS chemistry offers increased speed, greater fidelity, and longer reads. Furthermore, patterned flow cells and two-channel SBS have enhanced throughput and data accuracy [1].
Oxford Nanopore Sequencing
ONT technology takes a fundamentally different, real-time approach.
- Nanopore Sensing: A biological nanopore is embedded in a resistive membrane. As a single strand of DNA or RNA passes through the pore, it causes characteristic disruptions in an ionic current. Each nucleotide (or combination of nucleotides) produces a unique current signature, which is decoded into sequence data [3] [4].
- Key Features: This method allows for the direct sequencing of native DNA/RNA, enabling the detection of base modifications like methylation without special treatment [3]. It is also known for its portability and capacity for ultra-long reads.
The following diagram illustrates the core steps of the Illumina SBS workflow, highlighting the cyclical nature of its chemistry.
Performance Comparison: Key Metrics and Experimental Data
Direct comparative studies and platform specifications reveal distinct performance profiles for Illumina and Nanopore technologies, critical for experimental planning.
Accuracy and Error Profiles
- Illumina consistently demonstrates very high raw read accuracy, with a low error rate typically below 0.1% (Q30) [5]. This high base-calling accuracy is a result of the competitive incorporation of nucleotides and sophisticated image analysis [2]. For example, the iSeq 100 system maintains >80% of bases above Q30 (99.9% accuracy) even at 2x150 bp read lengths [6].
- Nanopore has historically had higher raw read error rates (5-15%), but recent advancements have led to significant improvements. The latest Q20+ chemistry and super-accuracy (SUP) basecalling can achieve single-read accuracy exceeding 99% [3]. However, its error profile is different, with a higher propensity for indels (insertions and deletions), especially in homopolymer regions, compared to Illumina's substitution-biased errors.
Table 1: Direct Platform Comparison in a Pathogen Identification Study [7]
| Metric | Illumina (MiSeq) | Oxford Nanopore (MinION) |
|---|---|---|
| Genus-level Concordance | 96.7% | 90.3% |
| Positive Predictive Value | 0.91 | 0.88 |
| Negative Predictive Value | 1.00 | 1.00 |
| Application Context | Identification of 31 bacterial pathogens from positive blood cultures using a Molecular Inversion Probe (MIP) panel. |
Table 2: Performance in 16S rRNA Microbiome Profiling [5]
| Metric | Illumina NextSeq (V3-V4) | ONT MinION (Full-Length) |
|---|---|---|
| Target Region | ~300 bp (V3-V4 hypervariable) | ~1500 bp (Full-length 16S) |
| Species Richness | Higher | Lower |
| Taxonomic Resolution | Genus-level | Species-level |
| Platform Bias | Detected broader range of taxa; overrepresented Prevotella, Bacteroides | Improved resolution for dominant species; overrepresented Enterococcus, Klebsiella |
| Application Context | 16S rRNA profiling of human and pig respiratory microbiomes. |
Read Length and Throughput
- Illumina generates short reads, typically up to 300 bp per end in paired-end runs (e.g., 2x300 bp on NextSeq) [5], or 2x250 bp on NovaSeq 6000 [8]. This is sufficient for many applications but can limit the resolution of complex genomic regions. Throughput is extremely high, with systems like the NovaSeq 6000 S4 flow cell producing up to 3000 Gb and 20 billion paired-end reads per run [8].
- Nanopore specializes in long reads, routinely producing reads tens of kilobases long, with ultra-long reads exceeding 4 Mb [4]. This is a key advantage for de novo assembly, resolving structural variants, and spanning repetitive regions. Throughput is scalable, from the portable Flongle and MinION to the high-output PromethION.
- Illumina Short-Reads:
- Strengths: Extremely high accuracy, high throughput, low cost per base, well-established and standardized workflows, excellent for variant calling and quantitative applications [4] [2].
- Limitations: Short read length limits ability to resolve complex structural variation, haplotyping, and repetitive regions; requires PCR amplification which can introduce bias.
- Nanopore Long-Reads:
- Strengths: Very long reads, real-time data analysis, portability, direct detection of base modifications, no PCR amplification required (for native DNA) [3] [4].
- Limitations: Historically higher error rates (though improving), higher raw read error rate can require more coverage for confident variant calling, throughput can be more variable.
Experimental Protocols for Technology Comparison
To ensure the data presented in the previous section is reproducible and transparent, this section details the key methodologies used in the cited comparative studies.
Protocol 1: Pathogen Identification via Molecular Inversion Probes (MIPs)
This protocol, used to generate the data in Table 1, highlights a probe-based targeted sequencing approach applicable to both platforms [7].
- 1. Probe Design & Sample Preparation: A MIP panel is designed to target pathogen-specific signatures. MIPs are single-stranded DNA probes that hybridize to the target, are extended by a DNA polymerase ("gap-filled"), and ligated to form a circular molecule. Genomic DNA is extracted from clinical samples (e.g., blood culture bottles, plasma).
- 2. MIP Capture Reaction: The DNA sample is incubated with the MIP pool, enzymes (DNA polymerase, ligase), and nucleotides. Circularized probes are purified using exonuclease digestion to remove linear, non-circularized DNA.
- 3. Universal Amplification: The captured target regions within the circularized MIPs are amplified using a single universal primer pair. The primer sequences are tailored for the specific sequencing platform (Illumina or ONT).
- 4. Library Preparation & Sequencing:
- For Illumina: Amplicons are barcoded using the Kapa HyperPlus kit and sequenced on a platform like MiSeq.
- For ONT: Amplicons are prepared using the Ligation Sequencing Kit and sequenced on a MinION.
- 5. Data Analysis: Reads are mapped to reference sequences for pathogen identification, and diagnostic metrics (concordance, predictive values) are calculated.
Protocol 2: Full-Length vs. Partial 16S rRNA Gene Sequencing
This protocol, underlying the data in Table 2, compares amplicon sequencing for microbiome analysis [5].
- 1. DNA Extraction: Genomic DNA is uniformly extracted from all samples (e.g., human and pig respiratory samples).
- 2. Library Preparation:
- For Illumina: The V3-V4 hypervariable region (~300 bp) of the 16S rRNA gene is amplified using specific primers (e.g., QIAseq 16S/ITS Region Panel). Libraries are sequenced on a NextSeq for 2x300 bp paired-end reads.
- For ONT: The full-length 16S rRNA gene (~1500 bp) is amplified and barcoded using the 16S Barcoding Kit (SQK-16S114.24). Libraries are sequenced on a MinION Mk1C with an R10.4.1 flow cell.
- 3. Bioinformatic Processing:
- Illumina Data: Processed using nf-core/ampliseq. Paired-end reads are quality filtered, error-corrected, and merged using DADA2 to generate Amplicon Sequence Variants (ASVs) for taxonomic classification.
- ONT Data: Basecalled and demultiplexed using Dorado. The EPI2ME Labs 16S Workflow is used for quality control and taxonomic classification against the SILVA database.
- 4. Downstream Analysis: Alpha and beta diversity metrics are calculated, and differential abundance analysis (e.g., with ANCOM-BC) is performed to identify platform-specific biases.
The workflow below visualizes the parallel paths taken in a direct comparative study, such as the 16S rRNA analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the protocols above relies on specific, high-quality reagents and materials. The following table details key solutions used in the featured comparative experiments.
Table 3: Key Research Reagent Solutions from Featured Experiments
| Item Name | Function / Description | Example Use Case |
|---|---|---|
| Molecular Inversion Probe (MIP) Panel | Single-stranded DNA probes for highly multiplexed targeted capture of genomic regions. | Simultaneous identification of dozens of bacterial, viral, and parasitic pathogens from a single sample [7]. |
| Ligation Sequencing Kit (SQK-LSK114) | Standard ONT library prep kit for genomic DNA; ligates adapters to dsDNA fragments. | Preparing amplicon or genomic DNA libraries for sequencing on MinION/PromethION flow cells [7] [9]. |
| QIAseq 16S/ITS Region Panel | A panel designed for targeted amplification of the 16S rRNA V3-V4 region for Illumina sequencing. | 16S rRNA microbiome profiling with Illumina short-read systems [5]. |
| 16S Barcoding Kit (SQK-16S114) | ONT kit for amplifying and barcoding the full-length 16S rRNA gene. | Full-length 16S sequencing for species-level taxonomic resolution [5]. |
| Agencourt AMPure XP Beads | Magnetic SPRI (Solid Phase Reversible Immobilization) beads for DNA size selection and purification. | Standard clean-up step in most NGS library preparation protocols, including MIP and ONT workflows [7] [9]. |
| PhiX Control Library | A well-characterized, sequencing-ready library used for quality control, alignment, and calibration. | Essential for quality monitoring and matrix calculation on Illumina sequencing runs [6] [8]. |
Illumina's short-read sequencing, built on its high-fidelity reversible terminator chemistry, remains the gold standard for applications demanding the highest base-level accuracy, such as single-nucleotide variant calling and quantitative gene expression in chemogenomics. In contrast, Oxford Nanopore sequencing provides a powerful complementary technology where long reads are paramount, including de novo assembly, structural variant detection, and direct epigenomic profiling. The choice is not necessarily one of superiority, but of fit-for-purpose. As the data shows, Illumina demonstrated a slight edge in concordance for targeted pathogen detection [7], while ONT enabled superior taxonomic resolution in microbiome studies via full-length 16S sequencing [5]. For the most comprehensive insights, a hybrid approach, leveraging the accuracy of Illumina with the long-range phasing of Nanopore, often represents the most robust strategy for complex chemogenomic research.
Next-generation sequencing (NGS) technologies have revolutionized genomic research, with Illumina and Oxford Nanopore Technologies (ONT) representing two fundamentally different approaches. Illumina sequencing utilizes synthesis-by-chemistry with fluorescently labeled nucleotides, generating short reads typically ranging from 100-300 base pairs with high per-base accuracy (exceeding 99.9%) [5] [10]. This technology excels in applications requiring high precision for single nucleotide variant detection but struggles with resolving repetitive regions and complex structural variations due to its short read length.
In contrast, Oxford Nanopore sequencing employs a novel physical approach based on the modulation of electrical currents as biomolecules pass through nanoscale pores. This technology produces long reads that can span thousands to millions of bases, enabling the resolution of complex genomic regions that remain challenging for short-read technologies [11]. A key advantage of nanopore sequencing is its ability to analyze native DNA and RNA without PCR amplification, allowing for direct detection of epigenetic modifications such as methylation alongside nucleotide sequence [3] [11].
The fundamental difference between these technologies extends beyond read length to their core biochemical principles. While Illumina relies on cyclic fluorescent imaging, nanopore technology transforms biological information into digital signals through changes in ionic current, creating unique opportunities and challenges for chemogenomic research applications [11].
The Fundamental Principles of Nanopore Technology
Core Sensing Mechanism
At the heart of Oxford Nanopore sequencing technology are biological nanopores embedded within an electro-resistant polymer membrane. Each nanopore corresponds to an individual electrode connected to a specialized sensor chip that measures the ionic current flowing through the pore [11]. The sensing process begins when a voltage is applied across this membrane, creating a constant ionic current flow through the nanopores as ions pass from one side to the other.
When DNA or RNA molecules are introduced to the system, they pass through these nanopores, causing characteristic disruptions in the electrical current. Each nucleotide base (A, T, G, C, or U) produces a distinctive disturbance pattern as it traverses the pore, resulting in what is known as a "squiggle" - the raw electrical signal that encodes the DNA or RNA sequence [12] [11]. This direct electrical detection method eliminates the need for PCR amplification, optical imaging, or chemical modification of the sample, enabling real-time analysis of native nucleic acids.
From Electrical Signals to Base Calling
The conversion of raw electrical signals to nucleotide sequences involves sophisticated machine learning algorithms that interpret the squiggle data. The basecalling software, such as Oxford Nanopore's Dorado, employs neural networks trained to recognize the distinctive current patterns associated with each nucleotide combination [13] [3]. This process occurs in real-time, allowing researchers to monitor sequencing progress and make dynamic decisions during experiments.
Recent advancements in basecalling algorithms have significantly improved accuracy through multiple processing modes:
- Fast basecalling: Optimized for speed and minimal computational requirements
- High Accuracy (HAC) mode: Balances accuracy with computational efficiency
- Super Accuracy (SUP) mode: Maximizes basecalling precision for demanding applications [3]
The latest basecalling models achieve raw read accuracies exceeding 99.75% (Q26), demonstrating substantial improvements over earlier versions of the technology [3].
Performance Comparison: Nanopore vs. Illumina
Accuracy and Error Profiles
Direct comparisons between Oxford Nanopore and Illumina sequencing reveal distinct error profiles and accuracy characteristics. Illumina sequencing consistently demonstrates higher raw read accuracy (99.68%, Q25) compared to Nanopore (96.84%, Q15), representing approximately a tenfold difference in error rates [10]. However, this discrepancy becomes less pronounced in consensus sequences, where Nanopore achieves Q50 (99.999%) accuracy at 10-20x coverage for bacterial assemblies [3].
Table 1: Sequencing Performance Metrics Comparison
| Parameter | Oxford Nanopore | Illumina |
|---|---|---|
| Raw Read Accuracy | 96.84% (Q15) to >99.75% (Q26) with latest chemistry [3] [10] | 99.68% (Q25) [10] |
| Typical Read Length | Hundreds to millions of bases [11] | 100-300 bp [5] |
| Consensus Accuracy | Q50 (99.999%) at 10-20x coverage [3] | Q25-Q30 (99.9-99.9%) [10] |
| Error Profile | Random errors across read length [5] | Higher toward read ends [5] |
| Epigenetic Modification Detection | Direct detection without special treatment [3] | Requires bisulfite conversion or other treatments [3] |
The error profiles between these technologies also differ significantly. Nanopore errors are typically randomly distributed across reads, while Illumina errors tend to cluster toward the 3' end of reads [5]. This distinction has important implications for downstream applications, with random errors being more readily correctable through consensus approaches.
Application-Specific Performance
The performance advantages of each technology vary considerably across different genomic applications:
16S rRNA microbiome profiling: Illumina captures greater species richness in complex microbial communities, while Nanopore provides superior species-level resolution due to its ability to sequence the full-length 16S rRNA gene (~1,500 bp) [5]. Taxonomic profiling reveals that Illumina detects a broader range of taxa, while Nanopore exhibits improved resolution for dominant bacterial species [5].
Whole genome assembly and structural variant detection: Nanopore excels in resolving repetitive regions and complex structural variations due to its long reads, achieving chromosome-scale haplotyping and enabling telomere-to-telomere assemblies [13] [3]. Illumina assemblies typically result in more fragmented genomes due to inability to span repetitive elements.
Transcriptome analysis: Nanopore long-read RNA sequencing enables full-length transcript characterization, allowing for precise identification of alternative isoforms, fusion transcripts, and RNA modifications [14]. Short-read RNA-seq struggles with transcript assembly and quantification of highly similar isoforms.
Epidemiological surveillance: For applications requiring high-resolution phylogenetic analysis, such as investigating transmission routes of bacterial pathogens, Illumina's higher accuracy provides more reliable single nucleotide polymorphism calls [10]. Nanopore offers advantages when rapid turnaround time is prioritized over ultimate resolution.
Table 2: Application-Based Performance Comparison
| Application | Nanopore Advantages | Illumina Advantages |
|---|---|---|
| Structural Variant Detection | Resolves complex regions and repetitive elements [3] [11] | Limited by short read length [11] |
| Metagenomic Classification | Species-level resolution with full-length 16S sequencing [5] | Greater species richness detection [5] |
| Transcript Isoform Analysis | Full-length transcript sequencing without assembly [14] | Requires complex transcript assembly [14] |
| Variant Calling | Phasing across long distances [3] | Higher SNP calling accuracy [10] |
| Epigenetic Analysis | Direct detection of base modifications [3] | Requires specialized treatments and protocols [3] |
Experimental Design and Methodologies
Representative Experimental Protocols
16S rRNA Microbiome Profiling Protocol
A comprehensive comparison of Illumina and Nanopore for respiratory microbiome analysis employed parallel processing of 34 respiratory samples from ventilator-associated pneumonia patients [5]. For Illumina sequencing, libraries targeted the V3-V4 hypervariable region (approximately 460 bp) using the QIAseq 16S/ITS Region Panel with 20 amplification cycles [5]. Sequencing was performed on the NextSeq platform to generate 2×300 bp paired-end reads.
For Nanopore sequencing, libraries were prepared using the ONT 16S Barcoding Kit 24 V14 (SQK-16S114.24) targeting the full-length 16S rRNA gene (~1,500 bp) [5]. Barcoded libraries were sequenced on MinION flow cells (R10.4.1) using MinKNOW software (v24.02.16) for up to 72 hours. Basecalling and demultiplexing used the Dorado basecaller (v7.3.11) with the High Accuracy model.
Bioinformatic processing followed platform-specific optimized pipelines: Illumina data used nf-core/ampliseq with DADA2 for amplicon sequence variant (ASV) calling, while Nanopore data used EPI2ME Labs 16S Workflow for taxonomic classification [5]. Both approaches utilized the Silva 138.1 prokaryotic SSU reference database.
Whole Genome Sequencing and Assembly Protocol
A comparison for bacterial pathogen surveillance sequenced 37 Clostridioides difficile isolates on both platforms [10]. Illumina libraries were prepared with the Nextera XT Kit and sequenced on NextSeq 500 with 2×150 bp reads, followed by quality control using Bifrost v1.1.1 and trimming with Trimmomatic v0.39 [10].
Nanopore libraries employed rapid barcoding kits (SQK-RBK110-96 and SQK-RBK114-96) sequenced on MinION devices with R9.4.1 and R10.4.1 flow cells [10]. Basecalling used Guppy v5.0.11 with super-accuracy mode, followed by adapter removal with qcat v1.1.0. Assembly approaches included Flye and Unicycler for Nanopore data, SPAdes for Illumina, and hybrid assembly approaches.
Technical Workflow Visualization
Workflow comparison between Nanopore and Illumina technologies
The Researcher's Toolkit: Essential Reagents and Materials
Core Sequencing Components
Successful implementation of nanopore sequencing requires specific reagents and materials optimized for the technology:
Flow Cells: Nanopore devices use specialized flow cells containing the nanopore array embedded in an electro-resistant membrane. Available formats include MinION (portable), PromethION (high-throughput), and GridION (mid-scale) flow cells [11].
Library Preparation Kits: Specific kits are available for different applications, such as the Ligation Sequencing Kit for genomic DNA, 16S Barcoding Kit for microbiome studies, and direct RNA sequencing kits for transcriptome analysis [5] [3].
Control Materials: Including known reference standards (e.g., human HG002 genome) for quality control and performance validation [3].
Basecalling Software: Dorado basecaller with multiple accuracy modes (Fast, HAC, SUP) for converting raw signals to nucleotide sequences [3].
Analysis Platforms: EPI2ME for user-friendly analysis workflows and MinKNOW for real-time run monitoring and control [13] [5].
Specialized Tools: Variant callers integrated with basecallers, modification detection algorithms, and assembly tools like Flye and Verkko optimized for long-read data [13] [3].
Future Directions and Emerging Applications
Technological Advancements
Oxford Nanopore continues to advance its technology platform, with recent developments focusing on increasing output and reducing costs. Current roadmap targets include 60-70% output enhancement into 2026, with a key milestone of 200 Gb per flow cell through chemistry improvements [13]. These advancements aim to lower the cost per genome, particularly for high-throughput human genomics applications.
The development of Q20+ chemistry with improved raw read accuracy exceeding 99% (Q20) represents another significant advancement, making nanopore sequencing competitive with short-read technologies for applications requiring high single-read accuracy [3]. Additionally, improvements in basecalling algorithms continue to enhance performance, with the latest Dorado models achieving 99.75% (Q26) raw read accuracy [3].
Expanding Application Horizons
The unique capabilities of nanopore sequencing are enabling new applications in chemogenomic research:
Single-molecule protein sequencing: Emerging research demonstrates the potential for nanopore-based detection of individual protein molecules, which could transform proteomic studies and clinical diagnostics [15].
Real-time targeted sequencing: Methods like UNCALLED and ReadUntil enable selective sequencing of genomic regions of interest by ejecting unwanted molecules from pores during sequencing, optimizing sequencing efficiency for targeted applications [16].
Integrated multiomic analysis: Nanopore's ability to sequence native DNA and RNA enables simultaneous detection of genetic sequence and epigenetic modifications, providing a more comprehensive view of genomic regulation [13] [17].
Portable sequencing solutions: The miniaturization of sequencing technology through devices like SmidgION aims to enable lab-free sequencing in field, clinical, and point-of-care settings [13].
These developments position nanopore sequencing as an increasingly versatile platform for chemogenomic research, with unique capabilities that complement rather than simply compete with Illumina short-read sequencing.
The field of DNA sequencing has undergone a remarkable transformation over the past decade, driven by competing technologies from Illumina and Oxford Nanopore Technologies (ONT). Within Illumina's ecosystem, the transition from the MiSeq to the NovaSeq platform represents a pursuit of unprecedented scale and throughput for large-scale genomic studies. Concurrently, Oxford Nanopore's evolution from the portable MinION to the high-output PromethION embodies a drive toward long-read sequencing at scale without sacrificing real-time analysis capabilities. For researchers in chemogenomic applications—where understanding the interaction between chemical compounds and biological systems is paramount—the choice between these platforms involves careful consideration of read length, accuracy, throughput, and application-specific requirements. This guide provides an objective comparison of these sequencing platforms, supported by recent experimental data, to inform strategic decisions in research and drug development.
The Illumina Evolution: MiSeq to NovaSeq
Illumina's sequencing platforms utilize sequencing-by-synthesis technology with reversible dye-terminators. The MiSeq system, with its maximum output of 8.5 Gb and 2 × 300 bp read length, has been a workhorse for targeted sequencing and amplicon applications [18]. The NovaSeq 6000 system represents a massive scaling up of this technology, capable of generating 20 billion paired-end reads (2400-3000 Gb) per run, making it suitable for large-scale whole-genome sequencing and population studies [18]. A significant difference lies in their flow cell chemistry: MiSeq uses a random lawn configuration while NovaSeq employs pre-defined binding spots for target DNA [18].
Table 1: Key Specifications of Illumina Sequencing Platforms
| Specification | MiSeq | NovaSeq 6000 | NovaSeq X Plus |
|---|---|---|---|
| Maximum Output | 7.5-8.5 Gb | 2400-3000 Gb | 3000-4000 Gb [19] |
| Reads per Run | Up to 50 million paired-end | Up to 20 billion paired-end | Not specified |
| Maximum Read Length | 2 × 300 bp | 2 × 150 bp | 2 × 150 bp |
| Run Time | 5-55 hours | 19-40 hours | Improved speed with v1.3 software [19] |
| Typical Applications | Targeted sequencing, 16S rRNA studies, small genomes | Whole-genome sequencing, large-scale transcriptomics, population studies | Large-scale genomics, multiomics, single-cell analysis [19] |
The Nanopore Evolution: MinION to PromethION
Oxford Nanopore's technology is based on measuring changes in ionic current as DNA or RNA molecules pass through protein nanopores. The MinION, a USB-powered portable device, enabled field sequencing and real-time analysis but with limited throughput. The PromethION platform represents a scaling of this technology for high-throughput projects, offering multiple flow cells that can be run independently or in parallel. A key advantage of Nanopore technology is its ability to generate ultra-long reads, with recent advances achieving reads exceeding 100 kb, which facilitates genome assembly and structural variant detection. The platform also allows for direct detection of DNA and RNA base modifications without specialized library preparation [20].
Table 2: Key Specifications of Oxford Nanopore Platforms
| Specification | MinION | PromethION |
|---|---|---|
| Read Length | Theoretical maximum >2 Mb; practical applications often 10-100 kb | Similar to MinION with capability for ultra-long reads |
| Throughput per Flow Cell | 10-30 Gb | 50-100 Gb per flow cell (varies by version) |
| Platform Size | USB-sized, portable | Benchtop instrument |
| Real-time Analysis | Yes, with live basecalling | Yes, with scalable compute options |
| Typical Applications | Field sequencing, rapid diagnostics, small genomes | Large genomes, transcriptomics, metagenomics |
Performance Comparison: Experimental Data
Throughput and Data Yield
Comparative studies demonstrate significant differences in data yield between platforms. In oral microbiome research, NovaSeq generated 193,081 ± 91,268 total reads compared to 71,406 ± 35,095 from MiSeq for the same samples—approximately 2.7 times more data [18]. NovaSeq also produced a higher percentage of high-quality, non-chimeric reads (45.25% vs. 41.78%), indicating not just greater quantity but superior quality in complex applications [18]. For Nanopore platforms, the PromethION enables scaling to whole human genomes at 40x coverage or higher in a single run, with raw signal data requiring approximately 1.7 TiB of storage for a human genome at 40x coverage [21].
Accuracy and Error Profiles
Error profiles differ substantially between short-read and long-read technologies. Illumina platforms typically exhibit error rates below 0.1% dominated by substitution errors [5]. In a direct comparison of 16S rRNA sequencing for respiratory microbiomes, Illumina demonstrated high accuracy for genus-level classification but struggled with species-level resolution due to shorter read lengths [5]. Nanopore sequencing has historically shown higher error rates (5-15%), though recent improvements in basecalling algorithms and flow cells (R10.4.1) have substantially improved accuracy [5] [22]. A 2025 study on Clostridioides difficile isolates reported Illumina read quality of 99.68% (Q25) compared to Nanopore at 96.84% (Q15)—approximately a tenfold difference in base-level accuracy [22].
Application-Specific Performance
Microbiome Studies
In oral microbiome research, NovaSeq detected significantly more unique operational taxonomic units (OTUs) compared to MiSeq, though community diversity metrics were similar between platforms [18]. For respiratory microbiome analysis, Illumina captured greater species richness, while ONT provided improved resolution for dominant bacterial species due to its longer reads [5]. ONT exhibited taxonomic biases, overrepresenting certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides) [5].
Whole Genome Sequencing
For bacterial genome assembly, Illumina's short reads result in highly accurate but fragmented assemblies, while Nanopore's long reads produce more contiguous genomes albeit with higher base-level errors [22]. Hybrid approaches that combine both technologies can leverage the advantages of each. In a C. difficile study, Nanopore sequencing alone was inadequate for high-resolution epidemiological surveillance due to an average of 640 base errors per genome but performed satisfactorily for detecting key virulence genes [22].
Transcriptomics
In RNA sequencing, long-read technologies excel at characterizing full-length transcript isoforms. A systematic benchmark of Nanopore long-read RNA sequencing demonstrated its superiority for identifying alternative isoforms, novel transcripts, fusion transcripts, and RNA modifications compared to short-read approaches [14]. The PCR-amplified cDNA Nanopore protocol requires the least input RNA and generates the highest throughput, while the direct RNA protocol enables sequencing of native RNA with base modification information [14].
Experimental Protocols for Platform Comparison
16S rRNA Sequencing Protocol for Microbiome Studies
Sample Preparation:
- DNA extraction from samples using standardized kits (e.g., Gram positive DNA purification kit)
- Quality assessment using PicoGreen and spectrophotometry
- Amplification of V1-V2 hypervariable regions using barcoded primers (27F: 5'-AGA GTT TGA TYM TGG CTC AG-3', 338R: 5'-TGC TGC CTC CCG TAG RAG T-3')
Library Preparation and Sequencing:
- Library preparation following Illumina 16S Metagenomic Sequencing Library protocols
- Equimolar pooling of amplicons
- Parallel sequencing on MiSeq and NovaSeq platforms [18]
- For Nanopore: Use ONT 16S Barcoding Kit following manufacturer's protocol [5]
Bioinformatic Analysis:
- Process reads using QIIME2 (for Illumina) or EPI2ME Labs 16S Workflow (for Nanopore)
- Calculate alpha diversity (Chao1, Shannon index) and beta diversity (Bray-Curtis)
- Taxonomic classification using Silva 138.1 database [18] [5]
Whole Genome Sequencing Protocol for Bacterial Isolates
DNA Extraction:
- Culture isolates under appropriate conditions (e.g., anaerobic for C. difficile)
- Perform mechanical or enzymatic lysis
- Purify DNA using commercial kits (e.g., MagNA Pure 96, DNeasy PowerSoil Pro Kit)
Library Preparation and Sequencing:
- For Illumina: Use Nextera XT Kit, sequence on NextSeq 500 with 2 × 150 bp reads
- For Nanopore: Use rapid barcoding kits (SQK-RBK110-96 or SQK-RBK114-96), sequence on MinION or PromethION
- For hybrid approaches: Prepare libraries for both platforms [22]
Data Analysis:
- Trim Illumina reads (Trimmomatic) and perform basecalling for Nanopore reads (Dorado)
- Assemble genomes using platform-specific (SPAdes for Illumina, Flye for Nanopore) or hybrid assemblers (Unicycler)
- Perform variant calling, phylogenetic analysis, and virulence gene detection [22]
Workflow Visualization
Diagram 1: Comparative sequencing workflow for Illumina and Nanopore platforms
Diagram 2: Data analysis pathways for short-read, long-read, and hybrid approaches
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Kits for Sequencing Platforms
| Item | Function | Platform Compatibility |
|---|---|---|
| Gram Positive DNA Purification Kit | DNA extraction from challenging samples | Both platforms [18] |
| Nextera XT DNA Library Preparation Kit | Library preparation for whole-genome sequencing | Illumina [22] |
| ONT 16S Barcoding Kit (SQK-16S114) | 16S rRNA amplification and barcoding | Oxford Nanopore [5] |
| Rapid Barcoding Kits (SQK-RBK110-96) | Quick library prep for multiplexing | Oxford Nanopore [22] |
| QIAseq 16S/ITS Region Panel | Targeted amplification of 16S regions | Illumina [5] |
| DNeasy PowerSoil Pro Kit | DNA extraction from soil and complex samples | Both platforms [22] |
| MagNA Pure 96 DNA and Viral NA Kit | Automated nucleic acid purification | Both platforms [22] |
The evolution from MiSeq to NovaSeq and from MinION to PromethION represents significant advancements in sequencing technology, each with distinct strengths for chemogenomic applications. Illumina's NovaSeq platform offers unparalleled throughput and base-level accuracy, making it ideal for large-scale studies requiring high statistical power, such as population genomics or drug response quantification. Oxford Nanopore's PromethION provides long reads and real-time analysis capabilities, advantageous for characterizing structural variants, transcript isoforms, and epigenetic modifications relevant to drug mechanisms.
Platform selection should be guided by specific research questions: NovaSeq excels in broad microbial surveys and large-scale genomic studies, while ONT provides superior resolution for complex genomic regions and rapid turnaround applications. Emerging hybrid approaches that leverage both technologies show promise for comprehensive genomic characterization in chemogenomic research. As both platforms continue to evolve—with Illumina's NovaSeq X Series offering enhanced multiomic capabilities and Oxford Nanopore improving basecalling accuracy and data compression—researchers now have powerful, complementary tools to advance drug discovery and development.
Next-generation sequencing (NGS) technologies are indispensable tools in modern chemogenomic research, enabling the high-throughput analysis required for drug discovery and microbial genomics. Among the available platforms, those developed by Illumina and Oxford Nanopore Technologies (ONT) represent two fundamentally different approaches. Illumina is renowned for its high-throughput and accuracy, while ONT offers long-read capabilities and real-time analysis. This guide provides a critical, objective comparison of their core technical specifications—read length, accuracy, throughput, and cost—framed within the context of chemogenomic applications. The analysis is supported by experimental data to help researchers and drug development professionals select the optimal technology for their specific research objectives.
The fundamental difference between Illumina and Nanopore technologies lies in their underlying sequencing biochemistry, which directly influences their performance characteristics.
Illumina employs sequencing by synthesis (SBS) chemistry. This method uses fluorescently labeled, reversible-terminator nucleotides. As DNA polymerase incorporates these nucleotides into the growing DNA strand, each base is identified by its specific fluorescent signal. This process occurs on flow cells containing millions of clusters, enabling massive parallel sequencing [23] [8]. This technology is the foundation for Illumina's high accuracy and throughput.
Oxford Nanopore technology is based on the electrical detection of nucleic acids. A biological nanopore is embedded in a membrane. As a single strand of DNA or RNA passes through the nanopore, it causes characteristic disruptions in an ionic current. These current changes are specific to the nucleotide sequence and are decoded in real-time to determine the DNA sequence [24]. This process does not require PCR amplification or labeled nucleotides, facilitating ultra-long reads and direct sequencing of native DNA or RNA.
The following workflow diagram illustrates the key procedural differences between the two platforms from sample to data analysis.
Comparative Technical Specifications
The choice between Illumina and Nanopore platforms requires a careful balance of their respective technical capabilities. The table below summarizes the core performance metrics for a direct comparison.
- Table 1: Core Technical Specification Comparison
| Specification | Illumina (Representative Models) | Oxford Nanopore (Representative Models) |
|---|---|---|
| Technology Principle | Sequencing by Synthesis (SBS) with fluorescent detection [23] [8] | Nanopore electrical current sensing [24] |
| Read Length | Short to Medium: Up to 2x300 bp (MiSeq) [23] | Very Long: Up to megabase-level fragments [24] |
| Single-Read Accuracy | Very High (Q30+): ~99.9% (error rate <0.1%) [5] [23] [25] | Moderate, Improving (Q20+): ~99% with latest V14 chemistry [26] [24] |
| Throughput per Run | Wide Range: 1.2 Gb (iSeq 100) to 3 Tb (NovaSeq 6000 S4) [6] [8] | Wide Range: Varies by device; PromethION up to 1.9 Tb [24] |
| Run Time | Hours to Days: ~4 hrs for 2x150 bp (MiSeq i100) to ~44 hrs for 2x150 bp (NovaSeq S4) [25] [8] | Hours to Days: Real-time data; run length is user-extendable (e.g., 72 hrs) [5] |
| Key Strength | High accuracy, high throughput, well-established bioinformatics | Long reads, real-time analysis, portability, direct RNA/epigenetic detection |
In-Depth Analysis of Specifications
Read Length: Illumina platforms generate short reads, typically up to 2x300 bp, which are sufficient for many applications like variant calling and gene expression counting [23]. In contrast, ONT produces long reads that can span entire 16S rRNA genes (~1,500 bp) or even megabase-long fragments, enabling the resolution of complex genomic regions, structural variations, and complete transcript isoforms [5] [24].
Accuracy: Illumina's main strength is its high per-base accuracy, typically exceeding Q30 (99.9% accuracy) [23] [25]. ONT has historically had higher error rates (5-15%), but recent advancements with the R10.4.1 flow cell and V14 chemistry (e.g., Kit14, Ligation Sequencing Kit V14) have significantly improved raw read accuracy to Q20 (99%) and above [5] [26] [10]. For applications requiring high consensus accuracy (e.g., genome assembly), both platforms can achieve >99.9% with sufficient coverage [24].
Throughput and Cost: Illumina offers a tiered ecosystem. The iSeq 100 provides low throughput (1.2 Gb) for small projects, the MiSeq i100 series offers mid-range output (up to 30 Gb), and the NovaSeq 6000 delivers ultra-high throughput (up to 3 Tb per flow cell) for population-scale studies [25] [6] [8]. ONT's throughput is more flexible, scaling from the portable MinION Mk1D to the benchtop PromethION 24 (up to 1.9 Tb) [26] [24]. ONT's initial instrument cost is often lower (MinION Mk1D at ~$5,000), while Illumina instruments represent a higher capital investment [26]. However, total cost per project must factor in consumables, which vary by application and scale.
Experimental Data and Performance Benchmarks
The theoretical specifications are best understood in the context of practical performance. Comparative studies across various genomic applications reveal distinct platform-specific biases and strengths.
16S rRNA Microbiome Profiling
A 2025 study compared Illumina (NextSeq, V3-V4 region) and ONT (MinION, full-length 16S) for respiratory microbiome analysis [5]. The experimental protocol involved collecting respiratory samples, extracting DNA, and performing parallel library preparation and sequencing on both platforms.
- Table 2: 16S rRNA Sequencing Performance [5]
| Metric | Illumina NextSeq | ONT MinION |
|---|---|---|
| Target Region | V3-V4 (~460 bp) | Full-length 16S (~1,500 bp) |
| Taxonomic Resolution | Reliable for genus-level classification | Enables species-level identification |
| Species Richness | Captured greater richness | Lower richness, but improved resolution for dominant species |
| Error Rate | Low (<0.1%) | Higher, but improved with latest chemistry |
| Key Finding | Ideal for broad microbial surveys | Excels in species-level resolution and real-time applications |
The study concluded that Illumina captured greater species richness, making it ideal for broad microbial surveys. ONT, with its longer reads, provided superior species-level resolution, making it better for identifying dominant bacterial species, though with some trade-offs in richness [5]. ANCOM-BC2 analysis further highlighted platform-specific biases, with ONT overrepresenting Enterococcus and Klebsiella while underrepresenting Prevotella and Bacteroides [5].
Whole-Genome Sequencing of Pathogens
Whole-genome sequencing (WGS) of bacterial pathogens is critical for tracking outbreaks and investigating antimicrobial resistance. A comparative study on Streptococcus pneumoniae demonstrated that newer ONT chemistry (R10.4.1 flow cells with Kit14) significantly improved the accuracy of MLST and antimicrobial resistance gene prediction compared to older versions, bringing its performance closer to that of Illumina [27]. Hybrid assembly, which combines long reads from ONT with short reads from Illumina, produced circular, high-quality genomes and is a recommended approach for generating complete reference sequences [27].
Conversely, a 2025 study on Clostridioides difficile highlighted the limitations of ONT-only data for high-resolution epidemiology. While ONT correctly identified virulence genes and sequence types (STs), its higher error rate (~96.84% accuracy, Q15) compared to Illumina (~99.68%, Q25) resulted in incorrect allele assignments in core-genome MLST (cgMLST) analysis. This made ONT-derived phylogenies less accurate for investigating fine-scale transmission events, though it was deemed suitable for rapid, less detailed analyses [10].
Essential Research Reagent Solutions
The experimental workflows for Illumina and ONT rely on specialized kits and reagents. The following table details key components used in the cited studies, providing a resource for experimental planning.
- Table 3: Key Research Reagents and Kits
| Item Name | Function / Description | Provider |
|---|---|---|
| QIAseq 16S/ITS Region Panel | Library preparation panel for targeted amplification of the 16S V3-V4 region for Illumina sequencing. | Qiagen [5] |
| Oxford Nanopore 16S Barcoding Kit 24 V14 (SQK-16S114.24) | Allows for amplification and full-length 16S rRNA sequencing with barcoding for up to 24 samples. | Oxford Nanopore [5] [26] |
| Nextera XT DNA Library Preparation Kit | Used for preparing Illumina sequencing libraries from bacterial genomic DNA via tagmentation. | Illumina [10] |
| SQK-LSK114 Ligation Sequencing Kit | A versatile ONT kit for genomic DNA sequencing, optimized for high accuracy (Q20+) with long reads. | Oxford Nanopore [26] |
| SQK-RBK114.96 Rapid Barcoding Kit | Enables simple and rapid library preparation with barcoding for up to 96 gDNA samples, reducing preparation time. | Oxford Nanopore [26] [10] |
| R10.4.1 Flow Cell | Nanopore flow cell with a dual-reader head design that improves basecalling accuracy, particularly in homopolymeric regions. | Oxford Nanopore [27] [10] |
The choice between Illumina and Oxford Nanopore Technologies is not a matter of one platform being superior to the other, but rather which is best suited to the specific goals of a chemogenomic research project.
Select Illumina when the research priority is maximum data accuracy and high throughput for applications such as variant calling, quantitative gene expression, and large-scale population studies where cost-per-base and reproducibility are critical. Its established protocols and bioinformatics tools make it a robust choice for standardized assays.
Select Oxford Nanopore when the research requires long-read sequencing, real-time analysis, or portability. ONT is the preferred technology for resolving complex genomic structures, performing full-length transcriptomics, direct detection of epigenetic modifications, and in-field or point-of-care sequencing where rapid turnaround is essential.
For the most comprehensive genomic characterization, a hybrid approach that leverages the high accuracy of Illumina short reads with the scaffolding power of ONT long reads often produces the highest-quality results, proving that these technologies are increasingly complementary in advancing chemogenomic research [27].
Chemogenomics represents a powerful, systematic approach in modern drug discovery that explores the interaction between chemical compounds and biological targets on a genome-wide scale. The core objective is to identify novel therapeutic targets and understand the mechanism of action of new chemical entities. The successful application of this strategy is fundamentally dependent on advanced genomic sequencing technologies, which provide the detailed molecular characterization required for target identification and compound profiling. Currently, two leading sequencing platforms, Illumina and Oxford Nanopore Technologies (ONT), dominate the research landscape, each offering distinct advantages and limitations.
Illumina sequencing is renowned for its exceptional accuracy and high throughput, making it a long-standing gold standard for applications requiring precise variant calling, such as whole-genome sequencing for rare variant discovery and genome-wide association studies (GWAS) [28] [29]. In contrast, Oxford Nanopore Technology is characterized by its long-read capabilities, real-time data analysis, and direct detection of epigenetic modifications like DNA methylation, without the need for pre-treatment [30] [31]. The choice between these platforms significantly influences the depth and quality of insights that can be gained in chemogenomic research. This guide provides an objective, data-driven comparison of their performance to inform researchers and drug development professionals in selecting the optimal technology for their specific applications.
Performance Comparison for Key Chemogenomic Applications
The utility of Illumina and Nanopore sequencing technologies varies significantly across different stages of the chemogenomics pipeline. The table below summarizes their performance in critical application areas, based on recent comparative studies.
Table 1: Platform Performance Across Core Chemogenomics Applications
| Application | Illumina (Short-Read) | Oxford Nanopore (Long-Read) | Supporting Evidence |
|---|---|---|---|
| Variant Discovery (SNPs/Indels) | High accuracy (Q25-Q30); Superior for common and rare variants [28]. | Higher error rates (~5-15%); requires polishing; Improved with latest basecallers [10] [5]. | WGS of 347,630 samples showed Illumina captured nearly 90% of heritability for 25 of 34 traits [28]. |
| Structural Variant & Complex Loci Analysis | Limited by short reads; struggles with repeats and homologous regions [29]. | Excels with long reads; resolves complex regions, repeats, and phasing [17]. | Enables haplotyping and parent-of-origin analysis (POAga) with 98% accuracy from a single sample [17]. |
| Epigenetic Profiling (Methylation) | Relies on bisulfite conversion, which degrades DNA and cannot distinguish 5mC from 5hmC [31]. | Direct, native detection of DNA methylation (5mC, 6mA) at single-base resolution without bisulfite conversion [31]. | Review highlights ONT's ability to natively detect methylation, unlike Illumina's bisulfite-seq [31]. |
| Metagenomics/ Microbiome Profiling | High sensitivity for species richness; ideal for broad microbial surveys (e.g., V3-V4 16S) [5]. | Species-level resolution with full-length 16S rRNA sequencing; rapid pathogen detection (<24 hrs) [30] [5]. | ONT identified 42 additional pathogens missed by standard methods in ICU samples [30]. |
| Transcriptomics (RNA Isoforms) | Indirect assembly of transcripts; challenges with alternative splicing, gene fusions, and isoform diversity. | Direct RNA sequencing captures full-length transcripts, enabling precise isoform identification and quantification [17]. | Single-cell long-read sequencing revealed Alzheimer's disease-specific isoform diversity [17]. |
| Portability & Turnaround Time | Lab-bound infrastructure; typical turnaround of days to weeks. | Portable (MinION); real-time data; results in hours for rapid diagnostics [10] [30]. | Study demonstrated pathogen ID in <24 hours, influencing antimicrobial therapy in 28% of cases [30]. |
Quantitative data from direct comparisons underscores a fundamental trade-off. A 2025 study on Clostridioides difficile sequencing found that Illumina produced reads with an average quality of 99.68% (Q25), while Nanopore reads reached 96.84% (Q15), showing a tenfold difference in raw accuracy [10]. This higher error rate in Nanopore data resulted in an average of 640 base errors per genome and incorrect assignment of over 180 alleles in cgMLST analysis, limiting its initial utility for high-resolution phylogenetic studies of transmission events [10]. Conversely, a comparative analysis of respiratory microbiomes found that while Illumina captured greater species richness, Nanopore's full-length 16S rRNA sequencing provided superior species-level resolution for dominant taxa, despite exhibiting biases in the relative abundance of certain genera [5].
Experimental Protocols and Methodologies
The divergent performance characteristics of Illumina and Nanopore platforms are rooted in their distinct underlying biochemical principles and experimental workflows. Reproducible results in chemogenomics depend on rigorous adherence to optimized, platform-specific protocols.
Illumina Workflow for Target Identification
The Illumina next-generation sequencing (NGS) workflow is a well-established process for target discovery.
Diagram 1: Core Illumina Sequencing Workflow
A typical Illumina workflow for whole-genome sequencing in target identification involves these critical steps [10] [29]:
- DNA Extraction: High-quality, high-molecular-weight genomic DNA is extracted from target cells or tissues. For transcriptomic analyses, RNA is extracted and converted to cDNA.
- Library Preparation: DNA is fragmented, often via sonication or enzymatic digestion, to a desired size (e.g., 300-800 bp). This is followed by end-repair, A-tailing, and ligation of platform-specific adapters. For complex disease research, hybridization-based capture (enrichment) can be applied to focus on exomes or specific gene panels [29].
- Cluster Generation: The adapter-ligated library is loaded onto a flow cell and undergoes bridge amplification, creating millions of clonal clusters, each representing a single template molecule.
- Sequencing: Sequencing-by-synthesis (SBS) occurs. Fluorescently labeled, reversible-terminator nucleotides are incorporated by DNA polymerase. After each incorporation, the flow cell is imaged, the fluorescent dye is cleaved, and the cycle repeats [31]. For methylation analysis, a bisulfite conversion step is required prior to library prep, which deaminates unmethylated cytosines to uracils [31].
- Data Analysis: The generated short reads are aligned to a reference genome. Downstream analysis for chemogenomics may include:
- Variant Calling: Using bioinformatic tools like DRAGEN to identify SNPs, indels, and copy number variations (CNVs) associated with disease or compound response [28].
- Differential Expression: For RNA-Seq, quantifying transcript abundance to identify genes dysregulated in disease or modulated by compound treatment [29].
- QTL Mapping: Correlating genetic variants with molecular phenotypes like gene expression (eQTLs) or methylation (meQTLs) to prioritize causal genes and pathways [29].
Nanopore Workflow for Comprehensive Profiling
The Nanopore sequencing workflow is notable for its simplicity and capacity for real-time analysis.
Diagram 2: Core Nanopore Sequencing Workflow
A standard Oxford Nanopore protocol for compound profiling involves [10] [30] [31]:
- Nucleic Acid Extraction: DNA or RNA is extracted, with a preference for long, intact molecules to leverage the long-read capability. Notably, RNA can be sequenced directly without conversion to cDNA.
- Library Preparation: This is a streamlined process. For DNA, the library is typically prepared by ligating a motor protein adapter directly to native DNA. The process does not require PCR amplification, thereby preserving base modifications. Adaptive sampling can be used for targeted sequencing of specific genomic regions of interest [17].
- Sequencing: The prepared library is loaded onto a flow cell containing nanopores embedded in an electro-resistant membrane. An ionic current is passed through the pores, and as DNA/RNA molecules are translocated through the pores by the motor protein, they cause characteristic disruptions in the current. These signal changes are specific to the nucleotide sequence and its modifications [31].
- Data Acquisition and Analysis: The raw current signals (squiggles) are converted to nucleotide sequences in real-time through a process called basecalling, which leverages sophisticated algorithms, often based on deep learning. This allows for immediate analysis. Key chemogenomic analyses include:
- Structural Variant Calling: Long reads enable precise mapping of large deletions, duplications, and translocations in cancer and rare disease genes [17].
- Direct Epigenetic Detection: Methylated cytosine (5mC) and other modifications are detected natively from the raw current signal, providing a simultaneous view of genetic and epigenetic variation from a single dataset [31].
- Full-Length Transcript Analysis: Direct RNA sequencing delivers accurate information on splice variants, fusion genes, and poly-A tail length, which is crucial for understanding compound effects on the transcriptome [17].
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting the appropriate consumables and bioinformatics tools is critical for the success of any sequencing project. The following table details essential solutions for implementing the described experimental protocols.
Table 2: Essential Research Reagent Solutions for Sequencing
| Item | Function/Description | Example Kits & Tools |
|---|---|---|
| High-Fidelity DNA Extraction Kit | Obtains high-molecular-weight, pure genomic DNA for long-read sequencing or complex library prep. | DNeasy PowerSoil Pro Kit [10], MagNA Pure 96 [10]. |
| Library Preparation Kit | Prepares nucleic acid fragments for sequencing by adding platform-specific adapters. | Illumina: Nextera XT [10]. Nanopore: Ligation Sequencing Kits, Rapid Barcoding Kits [10]. |
| Target Enrichment Solution | Enriches for specific genomic regions (e.g., exons, cancer panels) from complex genomes. | Illumina: 5-Base DNA Prep with Enrichment [32]. Nanopore: Adaptive Sampling (computational method) [17]. |
| Multiomic Sequencing Kit | Enables simultaneous detection of genomic and epigenomic information from a single library. | Illumina: 5-Base DNA Prep (uses proprietary conversion chemistry) [32]. |
| Bioinformatics Pipeline | Software for processing raw sequencing data, including basecalling, alignment, and variant calling. | Illumina: DRAGEN [28] [32]. Nanopore: Dorado basecaller, EPI2ME [5]. |
| Somatic Variant Caller | Specialized tool for identifying tumor-specific mutations by comparing to matched normal tissue. | DeepSomatic (optimized for long-read data, outperforms ClairS, Strelka2) [30]. |
The choice between Illumina and Oxford Nanopore Technologies for chemogenomics is not a matter of declaring a universal winner, but rather of aligning the technology's strengths with the specific research question.
Illumina sequencing remains the superior choice for applications where the highest possible accuracy is the primary determinant, such as discovering rare germline or somatic variants, conducting large-scale GWAS, and validating potential drug targets where base-level precision is non-negotiable [28]. Its high-throughput, cost-effective model is well-suited for massive population-level studies in early target discovery.
Oxford Nanopore sequencing is indispensable when the biological question requires context and comprehensiveness that short reads cannot provide. Its unique value lies in resolving complex genomic regions, identifying structural variants, phasing haplotypes, and directly detecting epigenetic marks and RNA modifications simultaneously with genetic sequence [17] [31]. This makes it powerful for elucidating complex mechanisms of action, understanding resistance mechanisms, and profiling the full spectrum of genomic variation in integrative multiomic studies.
For a comprehensive chemogenomics platform, a hybrid approach is often most powerful. Leveraging Illumina for its high accuracy in variant calling and Nanopore for its long-range phasing and epigenetic capabilities provides a synergistic strategy. This combined approach offers the most holistic view of the genome and its regulation, ultimately accelerating the identification and validation of novel therapeutic targets and the profiling of compound interactions.
Platform Applications in Antimicrobial Discovery, Microbiome Analysis, and Host Response
The rapid global spread of antimicrobial resistance (AMR) represents one of the most pressing public health challenges of our time, with projections estimating AMR could cause 10 million deaths annually by 2050 [33]. The effectiveness of antimicrobial treatments is increasingly compromised by the ability of bacteria to acquire and disseminate antimicrobial resistance genes (ARGs), particularly through mobile genetic elements (MGEs) like plasmids, transposons, and integrons [34] [35]. Understanding the precise genetic contexts and transmission mechanisms of ARGs is therefore critical for developing effective containment strategies.
Next-generation sequencing (NGS) technologies have revolutionized AMR research by enabling comprehensive genomic analysis. Among these platforms, Illumina short-read sequencing has served as the historical benchmark for AMR detection due to its high base-level accuracy (exceeding 99.9%) [36]. However, its limited read length (typically a few hundred base pairs) presents significant challenges for resolving repetitive regions and complex genomic structures where ARGs often reside [34] [37]. In contrast, Oxford Nanopore Technology (ONT) generates long reads that can span entire MDR regions and complex genetic structures, providing a more comprehensive view of the bacterial genome and enabling precise identification of ARGs and their associated MGEs [34] [33]. This capability is particularly valuable for studying the horizontal gene transfer of ARGs, a primary mechanism driving the rapid evolution and dissemination of resistance across diverse bacterial species [34] [35].
This guide provides an objective comparison of these two sequencing platforms, focusing specifically on their performance in resolving the genomic context of antimicrobial resistance genes and plasmids, a capability essential for understanding and combating the AMR crisis.
Technical Comparison of Sequencing Platforms
The fundamental differences in the underlying chemistry and data output between Illumina and Nanopore sequencing technologies directly impact their utility for AMR genomics.
Platform Mechanics and Data Output
Illumina technology utilizes sequencing-by-synthesis with reversible dye-terminators. Clonally amplified DNA fragments are sequenced in parallel on a flow cell, generating massive volumes of short reads with very high per-base accuracy [36]. This approach excels in detecting single nucleotide polymorphisms (SNPs) and variants with high confidence but struggles to resolve repetitive sequences and large structural variations due to fragmented assembly outcomes [34] [37].
Oxford Nanopore Technology (ONT) is based on the principle of passing single strands of DNA or RNA through a protein nanopore and measuring changes in electrical current as nucleotides translocate through the pore. This mechanism allows for the generation of ultra-long reads (N50 > 100 kb), real-time data analysis, and direct detection of DNA base modifications like methylation without additional processing [34] [37]. While historically associated with higher error rates, continuous improvements in nanopore proteins (e.g., the R10.4 flow cell with its dual reader head), motor enzymes, and base-calling algorithms have substantially improved raw read accuracy, now exceeding 99% with Q20+ chemistry [34].
Performance Metrics for AMR Applications
The table below summarizes the key performance characteristics of each platform relevant to AMR gene and plasmid analysis.
Table 1: Platform Performance Comparison for AMR Research
| Feature | Illumina (NextSeq) | Oxford Nanopore (MinION/PromethION) |
|---|---|---|
| Read Length | Short (up to 2x300 bp) [36] | Long (Ultra-long N50 > 100 kb) [34] |
| Typical Raw Read Accuracy | > 99.9% (Q30) [36] | ~99% (Q20) with latest chemistry [34] |
| Primary AMR Strength | High-confidence SNP/point mutation detection; broad microbial surveys [37] [36] | Resolving ARG context, plasmid structures, and horizontal gene transfer events [34] [33] |
| Assembly Outcome | Fragmented; poor resolution of repeats and MGEs [34] [37] | Highly contiguous; enables complete plasmid circularization [34] [35] |
| Turnaround Time | Hours to days (includes library prep and sequencing) | Real-time data analysis; rapid clinical resistance detection possible within hours [34] [38] |
| Portability | Benchtop instruments; limited mobility | Highly portable (MinION); suitable for field deployment [34] |
| DNA Modification Detection | Requires specialized library prep (e.g., bisulfite sequencing) | Direct, native detection of 5mC, 6mA, and 4mC from standard sequencing [37] |
Experimental Data and Comparative Analysis
Direct comparisons using real-world datasets demonstrate the practical implications of these technical differences for AMR research.
Resolving Antimicrobial Resistance Gene Contexts
A critical challenge in AMR surveillance is accurately linking resistance genes to their bacterial hosts and understanding their mobilization potential. Short-read sequencing often fails to assemble the repetitive flanks of MGEs, leading to incomplete and fragmented genomic context for ARGs [33]. In contrast, long nanopore reads can span entire resistance cassettes and operons, providing a complete picture of the genetic environment.
A comprehensive review analyzing 12 paired NGS-ONT datasets from municipal wastewater environments found that ONT significantly outperformed NGS in the assembly and identification of ARGs, MGEs, and plasmids. This advantage enables in-depth exploration of the co-occurrence between ARGs and MGEs, which is fundamental for assessing transmission risk [33]. Furthermore, a 2025 case study on fluoroquinolone resistance in chicken fecal samples leveraged ONT's ability to sequence native DNA and detect methylation patterns. Using tools like NanoMotif, researchers successfully linked an ARG-carrying plasmid to its bacterial host by identifying common DNA methylation signatures, a feat difficult to achieve with short-read data alone [37].
Table 2: Analysis of ARG and Plasmid Recovery in Wastewater Metagenomes (Adapted from [33])
| Genetic Element | Illumina Short-Read Assembly | Nanopore Long-Read Assembly | Implication for AMR Research |
|---|---|---|---|
| ARG Identification | High count but fragmented context | Improved contiguity reveals co-located ARGs | Uncovers potential for multi-drug resistance |
| Plasmid Reconstruction | Partial, often fragmented | High-quality, complete circular plasmids | Enables accurate tracking of plasmid spread and evolution |
| Mobile Genetic Elements (MGEs) | Poorly assembled due to repeats | Fully resolved structures (transposons, integrons) | Clarifies mechanisms of ARG horizontal transfer |
| Host Identification | Challenging for plasmids | Enabled via methylation binning and long-range linkage | Accurately identifies bacterial carriers of resistance |
Characterizing Plasmid Communities and Resistance Transmission
The ability to generate complete, circularized plasmid sequences is a standout strength of nanopore sequencing in AMR research. A seminal 2025 study investigated plasmids in wastewater treatment plant effluent, a known hotspot for AMR gene exchange. Using ONT, researchers sequenced and circularized 173 plasmids transferred into Escherichia coli. This revealed that 36% were mega-plasmids (>100 kb), and 73% of the AMR-positive plasmids were multidrug-resistant, carrying up to 12 different ARGs. Critically, the study found that plasmids predominantly existed as "communities" within a host cell, enabling non-AMR plasmids to survive antimicrobial selection by co-existing with resistant partners. This ecological insight into plasmid persistence was facilitated by the complete genomic context provided by long-read sequencing [35].
Detailed Experimental Workflow for Plasmid & ARG Analysis
To ensure reproducibility and provide a practical roadmap, here is a detailed protocol for analyzing AMR gene contexts and plasmids using nanopore sequencing, as implemented in recent studies [37] [38] [33].
Sample Preparation and Sequencing
- DNA Extraction: High-molecular-weight (HMW) genomic DNA is extracted from bacterial isolates or metagenomic samples using kits designed to preserve long DNA fragments (e.g., Norgen Biotek Sputum DNA Isolation Kit) [36]. DNA quality and fragment size are assessed via fluorometry (Qubit) and pulsed-field gel electrophoresis or TapeStation.
- Library Preparation: For ONT, the HMW DNA is prepared using a ligation sequencing kit (e.g., SQK-LSK114). A key advantage is the ability to sequence native DNA without PCR amplification, allowing for direct detection of base modifications. The library is loaded onto a flow cell (R10.4.1 or newer is recommended for improved homopolymer accuracy) [34] [37] [36].
- Sequencing: Sequencing is performed on a MinION, GridION, or PromethION platform using MinKNOW software. Data acquisition can run for up to 72 hours or until the flow cell is exhausted, with basecalling performed in real-time using the Dorado basecaller with a High Accuracy (HAC) model [37] [36].
Bioinformatic Analysis
The following workflow outlines the primary steps for data processing, from raw signals to biological insights.
Diagram 1: Bioinformatic workflow for nanopore-based AMR analysis.
- Basecalling and Demultiplexing: Convert raw current signals (
FAST5) to nucleotide sequences (FASTQ) and separate reads by sample barcode using ONT's Dorado or Guppy software [36] [33]. - Quality Control and Filtering: Remove adapters and filter out low-quality reads (e.g., quality score < 7, length < 1000 bp) using tools like Porechop and Nanofilt. General statistics and visualizations are generated with NanoPlot [33].
- Genome Assembly: Perform de novo assembly using long-read specific assemblers such as Flye or Canu to generate highly contiguous sequences and circularize plasmids [33] [35].
- Gene Annotation and Identification: Annotate assembled contigs using tools like Prokka. Identify ARGs and MGEs by comparing contigs and/or raw reads against specialized databases such as the Comprehensive Antibiotic Resistance Database (CARD) using ABRicate or ARGpore [38] [33].
- Advanced Analysis: Methylation-Based Host Linking: For metagenomic samples, use tools like MicrobeMod or NanoMotif to detect DNA methylation motifs (6mA, 5mC, 4mC) from the sequencing data. Plasmids and chromosomes from the same host will share a common methylation pattern, allowing for accurate binning and host assignment [37].
Successful implementation of nanopore sequencing for AMR research relies on a suite of specialized wet-lab and bioinformatic tools.
Table 3: Key Reagents and Resources for Nanopore AMR Analysis
| Item | Function/Description | Example Products/Tools |
|---|---|---|
| HMW DNA Extraction Kit | Isolates long, intact DNA fragments crucial for long-read sequencing. | Norgen Biotek Sputum DNA Isolation Kit [36] |
| ONT Ligation Sequencing Kit | Prepares genomic DNA libraries for sequencing on Nanopore devices. | ONT SQK-LSK114 Ligation Sequencing Kit [37] |
| Flow Cell | The consumable containing nanopores for sequencing. | ONT R10.4.1 flow cell (for improved accuracy) [37] [36] |
| Basecaller | Software that translates raw electrical signals into DNA sequences. | Dorado basecaller (High Accuracy model) [36] |
| Long-Read Assembler | Assembles long reads into contiguous sequences (contigs). | Flye, Canu [33] |
| ARG Database | Curated database of reference sequences for identifying ARGs. | Comprehensive Antibiotic Resistance Database (CARD) [38] [33] |
| Methylation Analysis Tool | Detects DNA base modifications and links plasmids to hosts. | NanoMotif, MicrobeMod [37] |
The choice between Illumina and Nanopore sequencing for antimicrobial resistance research is not a matter of identifying a universally superior technology, but rather of selecting the right tool for the specific research question. For comprehensive, high-throughput detection of known resistance determinants and point mutations across large sample sets, Illumina's high accuracy remains a powerful choice. However, for investigations demanding a complete understanding of ARG transmission, plasmid ecology, and the complex genetic contexts driving resistance spread, Oxford Nanopore's long-read technology offers transformative capabilities.
The capacity of nanopore sequencing to fully resolve plasmid structures, link them to their bacterial hosts via methylation patterns, and operate in real-time positions it as an indispensable technology for advanced AMR surveillance and outbreak response. As sequencing costs continue to decrease and analytical pipelines become more robust, the integration of long-read data is poised to become standard practice in the ongoing global effort to combat antimicrobial resistance.
The human microbiome plays a crucial role in drug metabolism and efficacy, making accurate microbial community analysis essential for chemogenomic research. Two principal sequencing methods dominate this field: 16S rRNA gene amplicon sequencing and shotgun metagenomic sequencing. The 16S rRNA approach targets the amplification and sequencing of specific variable regions of the bacterial 16S ribosomal RNA gene, providing a cost-effective method for taxonomic classification primarily at the genus level [39] [40]. In contrast, shotgun metagenomics sequences all the DNA present in a sample, enabling not only species-level taxonomic resolution but also functional profiling of microbial communities [39] [41]. Within the context of chemogenomic applications, the choice between these methods significantly impacts the depth of information available for understanding microbe-drug interactions.
The ongoing evolution of sequencing technologies further complicates this choice, primarily between Illumina's short-read platforms and Oxford Nanopore Technologies' (ONT) long-read capabilities. Illumina sequencing provides high accuracy for short reads and remains the workhorse for both 16S studies and shotgun metagenomics [42] [43]. Meanwhile, Nanopore technology generates long reads that can span the entire ~1.5 kb 16S rRNA gene in a single read or produce contiguous assemblies in shotgun approaches, overcoming fragmentation issues and enabling more accurate strain-level discrimination [44] [45]. This technical comparison is particularly relevant for drug response studies where identifying functional genes and metabolic pathways is paramount.
Technical Comparison of 16S rRNA and Shotgun Metagenomics
Key Characteristics and Trade-offs
16S rRNA sequencing offers a targeted, cost-effective approach for bacterial community profiling. By focusing on specific hypervariable regions (e.g., V3-V4 or V4-V5) of the conserved 16S rRNA gene, this method requires fewer sequencing reads per sample (approximately 50,000 reads) to maximize taxon identification [40]. However, its reliance on a single gene marker introduces several limitations: restricted taxonomic resolution (typically to genus level), inability to profile non-bacterial community members (viruses, fungi, eukaryotes), and dependence on PCR amplification which can introduce sequence artifacts and quantification biases [39] [40]. Additionally, the choice of primer pairs and target regions significantly affects community characterization, and the method provides only indirect functional inference through predictive tools [40].
Shotgun metagenomics employs untargeted sequencing of all DNA in a sample, providing several advantages for comprehensive microbiome analysis. This approach enables species-level taxonomic assignment and can detect non-bacterial microorganisms, including viruses and fungi [39] [40]. Most importantly for drug response studies, shotgun sequencing allows direct characterization of functional genes and metabolic pathways, including those involved in drug metabolism [40] [41]. The main disadvantages include substantially higher sequencing depth requirements (millions of reads per sample), increased costs, and heavier computational demands for data analysis [40]. Unlike 16S sequencing, shotgun metagenomics relies heavily on reference databases, which can limit detection of novel microbes without computationally expensive assembly procedures [40].
Table 1: Technical Comparison of 16S rRNA and Shotgun Metagenomic Sequencing
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Sequencing Target | Specific variable regions of 16S rRNA gene | All genomic DNA in sample |
| Taxonomic Resolution | Genus-level (sometimes species) | Species to strain-level |
| Organisms Detected | Bacteria primarily | Bacteria, viruses, fungi, archaea |
| Functional Profiling | Indirect prediction | Direct assessment of genes and pathways |
| PCR Amplification Bias | Yes | No (for DNA-based approaches) |
| Sequencing Depth Required | ~50,000 reads/sample | Millions of reads/sample |
| Cost per Sample | Lower | Higher |
| Reference Database Dependency | Moderate | High |
| Bioinformatics Complexity | Moderate | High |
Performance in Microbial Diversity Assessment
Multiple studies have directly compared the performance of 16S rRNA and shotgun metagenomic sequencing for diversity assessment. In pediatric gut microbiome studies, both methods identified similar patterns of alpha diversity (within-sample diversity) and beta diversity (between-sample differences), despite detecting different numbers of taxa [40] [43]. Notably, 16S rRNA profiling often identifies a larger number of genera, while shotgun sequencing provides more accurate species-level identification [40].
A comparative analysis of migratory seagull gut microbiota found that the correlation between methods gradually decreased with refinement of taxonomic levels [39]. The largest differences in relative abundance occurred at the species level, where shotgun metagenomics identified many pathogenic bacteria that were missed by 16S sequencing [39]. Both methods showed high consistency in beta diversity metrics at the genus level, suggesting comparable patterns of microbial community structure despite technical differences [39].
In clinical diagnostics, shotgun metagenomics demonstrated significantly better performance for bacterial detection at the species level compared to 16S rRNA Sanger sequencing [46]. One prospective study found shotgun metagenomics identified a bacterial etiology in 46.3% of cases versus 38.8% with 16S sequencing, with the difference becoming particularly significant at the species level (28/67 vs. 13/67) [46].
Experimental Data and Benchmarking Studies
Comparative Performance Across Sequencing Platforms
Recent benchmarking studies have evaluated multiple sequencing technologies for microbiome analysis. One comprehensive study compared seven sequencing platforms encompassing both second-generation (Illumina HiSeq 3000, MGI DNBSEQ-G400, DNBSEQ-T7, ThermoFisher Ion GeneStudio S5, Ion Proton P1) and third-generation sequencers (Oxford Nanopore MinION, Pacific Biosciences Sequel II) using complex synthetic microbial communities [42].
Table 2: Performance Metrics Across Sequencing Technologies for Mock Microbial Communities
| Sequencing Technology | Read Length (bp) | % Mapped End-to-End | % Uniquely Mapped | % Avg Identity | Key Strengths |
|---|---|---|---|---|---|
| Illumina HiSeq 3000 | 149 ± 4.24 | 99.62% | 93.21% | 99.45% | High accuracy, established protocols |
| Ion Proton P1 | 144.04 ± 28.43 | 99.29% | 87.13% | 99.42% | Rapid turnaround |
| ONT MinION R9 | 4408.41 ± 2831.95 | 99.75% | 99.63% | 89.08% | Long reads, real-time analysis |
| PacBio Sequel II | 10289.7 ± 4036.27 | 99.65% | 99.62% | 99.72% | Very long reads with high accuracy |
| DNBSEQ-G400 | 99.91 ± 0.96 | 99.22% | 89.16% | 99.70% | Low indel rate, cost-effective |
The study demonstrated that while third-generation sequencers like Oxford Nanopore and PacBio have advantages for analyzing complex microbial communities, they require careful library preparation for optimal quantitative metagenomic analysis [42]. Notably, long-read technologies achieved almost 100% uniquely mapped reads, significantly higher than most short-read platforms [42]. However, the Oxford Nanopore MinION R9 showed lower read identity (89.08%) due to higher indel and substitution errors compared to other technologies [42].
Bioinformatics Pipeline Performance
The choice of bioinformatics pipeline significantly impacts taxonomic classification accuracy in shotgun metagenomics. A recent assessment of publicly available shotgun metagenomic processing packages used 19 mock community samples to evaluate performance [41].
Table 3: Performance Metrics of Shotgun Metagenomics Bioinformatics Pipelines
| Pipeline | Classification Approach | Key Features | Performance Notes |
|---|---|---|---|
| bioBakery (MetaPhlAn4) | Marker gene + MAG-based | Uses known/unknown species-level genome bins | Best overall performance in accuracy metrics |
| JAMS | Kraken2 k-mer based | Includes genome assembly | High sensitivity |
| WGSA2 | Kraken2 k-mer based | Optional genome assembly | High sensitivity |
| Woltka | Operational Genomic Unit (OGU) | Phylogeny-based, uses evolutionary history | Newer approach with potential for refined classification |
The benchmarking revealed that bioBakery4 performed best for most accuracy metrics, while JAMS and WGSA2 achieved the highest sensitivities [41]. This comprehensive assessment highlights the importance of pipeline selection in shotgun metagenomic analysis, particularly for drug response studies where accurate taxon identification and quantification are critical.
Methodological Protocols
Standardized 16S rRNA Sequencing Workflow
The 16S rRNA sequencing workflow involves several critical steps to ensure reproducible results. For Illumina platforms, the typical protocol targets hypervariable regions (e.g., V3-V4 or V4-V5) of the 16S rRNA gene. Samples are first pre-treated to lyse human cells and degrade human nucleic acids, followed by specialized DNA extraction using kits such as the ZymoBIOMICS DNA Miniprep Kit for environmental samples or QIAamp PowerFecal DNA Kit for stool samples [44] [43]. The target region is then amplified using PCR with barcoded primers (e.g., 515FB/806RB for the V4 region), and libraries are prepared using kits such as the Illumina MiSeq Reagent Kit [43]. Sequencing is typically performed on Illumina MiSeq or NovaSeq systems with 2×150bp or 2×250bp paired-end reads [39] [43].
For Oxford Nanopore platforms, the 16S Barcoding Kit enables amplification of the entire ~1.5 kb 16S rRNA gene using barcoded primers, followed by sequencing adapter addition [44]. This approach sequences the entire gene rather than subsets, achieving higher taxonomic resolution for accurate species identification from polymicrobial samples [44] [45]. Libraries are typically sequenced on MinION or GridION devices using the high-accuracy (HAC) basecaller, with flow cells potentially reused multiple times using Wash Kits to reduce costs [44]. The EPI2ME wf-16s pipeline provides automated analysis, generating abundance tables and interactive visualizations [44].
Shotgun Metagenomics Sequencing Workflow
Shotgun metagenomics employs a fundamentally different approach that begins with comprehensive DNA extraction from all organisms in a sample. For Illumina-based workflows, extracted DNA is fragmented to appropriate sizes (typically 350bp), followed by end-repair, A-tailing, and adapter ligation using kits such as the NEB Next DNA Library Prep Kit [39]. DNA fragments of 300-400bp are enriched by PCR and purified using an AMPure XP system [39]. Libraries are quantified and sequenced on Illumina NovaSeq or NextSeq systems with paired-end 150bp reads [39] [43]. Critical quality control steps include host-derived read removal using tools like KneadData and quality filtering with Trim_Galore [43].
For Oxford Nanopore shotgun metagenomics, library preparation avoids fragmentation to preserve long reads, using kits such as the Rapid Ligation Kit [47]. Sequencing occurs on MinION, GridION, or PromethION platforms, with PromethION 2 devices particularly recommended for obtaining complete, circular metagenome-assembled genomes (MAGs) from complex microbial samples [48] [47]. Recent advancements in nanopore sequencing, including kit 14 and R10 chemistry, have significantly improved accuracy, with new bioinformatics tools like nanoMDBG enabling efficient construction of hundreds of high-quality MAGs from challenging sample types [47].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Kits for Microbiome Profiling
| Category | Product/Kit | Application | Key Features |
|---|---|---|---|
| DNA Extraction | QIAamp PowerFecal DNA Kit (Qiagen) | Stool microbiome DNA extraction | Optimized for difficult-to-lyse microbes, inhibitor removal |
| ZymoBIOMICS DNA Miniprep Kit | Environmental water samples | Maintains community representation | |
| QIAGEN DNeasy PowerMax Soil Kit | Soil samples | Effective for humic acid removal | |
| 16S Library Prep | Illumina MiSeq Reagent Kit | 16S amplicon sequencing | Standardized workflow for V3-V4 regions |
| Oxford Nanopore 16S Barcoding Kit | Full-length 16S sequencing | Multiplexes 24 samples, sequences V1-V9 regions | |
| Shotgun Library Prep | Nextera XT DNA Library Prep Kit (Illumina) | Shotgun metagenomics | Tagmentation-based, fast workflow |
| Oxford Nanopore Rapid Ligation Kit | Long-read metagenomics | Preserves long fragments, minimal bias | |
| Analysis Tools | DADA2 (16S) | Amplicon sequence variant analysis | Denoises, removes chimeras, genus-level assignment |
| MetaPhlAn4 (Shotgun) | Taxonomic profiling | Marker-based, integrates MAGs | |
| nanoMDBG (ONT) | Metagenome assembly | Optimized for nanopore data, efficient MAG construction |
Application in Drug Response Studies
The choice between 16S rRNA and shotgun metagenomic sequencing has profound implications for chemogenomic research into drug-microbiome interactions. Shotgun metagenomics provides the comprehensive functional profiling necessary for understanding microbial drug metabolism, identification of resistance genes, and discovery of microbial bioactive compounds [40] [41]. The ability to reconstruct metagenome-assembled genomes (MAGs) from shotgun data enables researchers to link specific metabolic capabilities to individual microbial strains, crucial for personalized medicine approaches [48] [47].
For longitudinal studies tracking microbiome changes during drug treatment, both methods can detect shifts in microbial diversity, but shotgun sequencing offers superior resolution for identifying specific functional changes at the genetic level [40] [43]. However, 16S rRNA sequencing remains valuable for large-scale cohort studies where cost considerations limit deep sequencing, and where taxonomic profiling at genus level provides sufficient information [40] [43]. Research has demonstrated that 16S rRNA data can achieve similar predictive accuracy for disease states compared to shotgun data, with area under the receiver operating characteristic curve (AUROC) values of approximately 0.90 for both methods in pediatric ulcerative colitis classification [43].
The integration of long-read technologies from Oxford Nanopore is particularly promising for drug response studies, as it enables real-time sequencing and analysis of complex microbial communities without the need for culturing [45] [47]. Recent advances have demonstrated that nanopore sequencing can now produce results comparable to PacBio HiFi sequencing at equivalent depths, while providing the additional advantage of rapid pathogen identification and antimicrobial resistance profiling [47]. This capability for rapid, in-house analysis of microbiome samples has significant implications for clinical drug development and personalized treatment strategies.
The discovery of robust bacterial biomarkers for diseases like colorectal cancer (CRC) and head and neck cancer (HNC) is a pivotal goal in modern medical research. For years, short-read sequencing of partial 16S rRNA gene regions (e.g., V3-V4) on Illumina platforms has been the standard, providing reliable genus-level community profiles. However, the limited resolution of this approach often obscures the specific species and strains that drive disease pathophysiology. The advent of third-generation sequencing, specifically Oxford Nanopore Technologies (ONT) and its capability for full-length 16S rRNA sequencing, promises to overcome this limitation. This guide objectively compares the performance of full-length 16S ONT sequencing against Illumina V3-V4 sequencing, focusing on their utility for bacterial biomarker discovery, supported by recent experimental data.
Technical Comparison: Platform Capabilities and Performance
The fundamental differences between the two sequencing technologies dictate their performance in microbiome analysis.
Table 1: Core Technical Specifications of Sequencing Platforms for 16S rRNA Analysis
| Feature | Illumina (V3-V4) | Oxford Nanopore (Full-Length V1-V9) |
|---|---|---|
| Read Length | Short reads (~300-500 bp) [49] | Long reads (>1,500 bp, full-length gene) [50] |
| Target Region | Select hypervariable regions (e.g., V3-V4) | All nine hypervariable regions (V1-V9) |
| Typical Taxonomic Resolution | Primarily genus-level [49] [51] | Species-level and often strain-level [50] [52] |
| Primary Strength | High throughput, low per-base error rate (~0.1%) [5] | Superior taxonomic resolution, real-time analysis, lower capital cost [50] |
| Primary Limitation | Limited species-level resolution due to short read length [52] | Historically higher error rates, though significantly improved with R10.4.1 chemistry and Dorado basecallers [50] [5] |
Key Findings from Comparative Studies
Direct comparisons across multiple sample types consistently demonstrate the advantage of full-length sequencing for detailed taxonomic classification.
Species-Level Identification and Validation
In head and neck cancer tissues, full-length ONT sequencing demonstrated a four-fold increase in species-level identification accuracy compared to V3-V4 Illumina sequencing. When validated against culture-based identification using MALDI-TOF MS, ONT correctly identified 75% of bacterial isolates at the species level, while Illumina managed only 18.8% [49] [51] [53]. This establishes the superior real-world accuracy of ONT for species-specific biomarker discovery.
Biomarker Discovery in Colorectal Cancer
A large-scale study on fecal samples from 123 subjects revealed that Nanopore full-length 16S sequencing identified a wider and more specific array of bacterial biomarkers for colorectal cancer compared to Illumina V3-V4 [50]. The species-level resolution of ONT facilitated the construction of a predictive model for CRC with an AUC (Area Under the Curve) of 0.87 using 14 species, and an AUC of 0.82 with just 4 key species (Parvimonas micra, Fusobacterium nucleatum, Bacteroides fragilis, and Agathobaculum butyriciproducens) [50].
Diversity Metrics and Community Composition
Studies generally report that alpha diversity (within-sample diversity) is similar between the two techniques [49] [54]. However, beta diversity (between-sample diversity) often shows significant differences, indicating that each technique can capture a distinct aspect of the microbial community structure [49] [5]. Correlation in microbial relative abundance between platforms is high at the phylum and family levels but decreases substantially at the species level, underscoring the unique resolution of the full-length approach [49] [51].
Table 2: Summary of Comparative Performance from Recent Studies
| Study & Sample Type | Species-Level ID (ONT vs. Illumina) | Key Findings for Biomarker Discovery |
|---|---|---|
| Head and Neck Cancer Tissues [49] | 75% vs. 18.8% (vs. MALDI-TOF MS) | FL-ONT provides significantly better resolution at lower taxonomic levels. |
| Colorectal Cancer Feces [50] | Superior species-level resolution | ONT identified specific CRC biomarkers (e.g., F. nucleatum, P. micra) and enabled high-AUC predictive models. |
| Respiratory (Tracheal Aspirates) [54] | Superior species-level resolution | FL-ONT with Emu pipeline provided comparable diversity metrics but superior species-level resolution for pathogenic genera. |
Detailed Experimental Protocols from Cited Studies
To ensure reproducibility and provide a clear technical context for the data presented, here are the detailed methodologies from key studies.
- Sample Collection: Tumor tissues from 26 HNC patients were collected in sterile cryotubes immediately after surgical resection.
- DNA Extraction: Tissues were homogenized with steel beads, enzymatically treated with lysozyme, lysostaphin, and proteinase K, and DNA was purified using the DNeasy Blood & Tissue Kit (Qiagen).
- V3V4-Illumina Sequencing: PCR amplification was performed using V3-V4 primers (341F, 806R). Sequencing was conducted on an Illumina platform by the Australian Genome Research Facility.
- FL-ONT Sequencing: The full-length V1-V9 region was amplified with 27F and 1492R primers. Libraries were prepared using the 16S Barcoding Kit (SQK-16S024) and sequenced on a MinION flow cell (R9.4.1).
- Bioinformatic Analysis: Illumina data was processed with DADA2 and QIIME2. ONT data was processed using the Emu pipeline for taxonomic assignment.
- Cohort: 123 subjects (93 CRC patients, 30 healthy controls).
- DNA Extraction & Library Prep: Fecal DNA was extracted. For Illumina, the V3-V4 region was amplified and sequenced. For ONT, the full-length V1-V9 region was amplified and libraries were prepared with the 16S Barcoding Kit.
- Sequencing: ONT sequencing was performed on R10.4.1 flow cells using the Dorado basecaller (with fast, hac, and sup models).
- Bioinformatic Analysis: Illumina data was processed with DADA2 and QIIME2. ONT data was analyzed with the Emu pipeline, using both SILVA and Emu's default database for taxonomic classification.
- Sample Processing: Tracheal aspirates were processed using multiple DNA extraction kits (QIAamp BiOstic, MagMax, Zymo HostZero) to evaluate bias.
- Sequencing: Both Illumina V3-V4 and ONT FL-16S (using 27F-1492R primers) sequencing were performed.
- Bioinformatic Analysis: Illumina data was processed with QIIME2. ONT FL-16S reads were analyzed using the Emu pipeline, which is specifically designed for the error profile of long reads and enhances classification accuracy.
Visualizing the Experimental Workflow
The following diagram illustrates the typical parallel workflow for a comparative study of the Illumina V3-V4 and ONT full-length 16S sequencing approaches.
Comparative 16S rRNA Sequencing Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for 16S rRNA Sequencing Studies
| Item | Function | Example Use Case |
|---|---|---|
| DNeasy Blood & Tissue Kit (Qiagen) | DNA extraction from complex samples like tissue. | Used for extracting DNA from head and neck cancer tumor tissues [49]. |
| QIAseq 16S/ITS Region Panel (Qiagen) | Targeted amplification and library prep for Illumina. | Used for sequencing the V3-V4 region on the Illumina NextSeq platform [5]. |
| ONT 16S Barcoding Kit (SQK-16S024) | PCR-based library prep for full-length 16S on Nanopore. | Used for amplifying the V1-V9 region for sequencing on MinION flow cells [49] [55]. |
| R10.4.1 Flow Cell (ONT) | Nanopore flow cell with improved accuracy. | Key for achieving high-quality, full-length 16S reads with lower error rates [50]. |
| ZymoBIOMICS Microbial Standards | Defined mock community for protocol validation. | Used as a positive control to assess extraction and sequencing bias [56]. |
For broad microbial community profiling where genus-level information is sufficient, Illumina V3-V4 sequencing remains a powerful and cost-effective tool. However, for discovering disease-specific bacterial biomarkers, the evidence strongly supports the adoption of full-length 16S rRNA sequencing with Oxford Nanopore. The species-level resolution provided by ONT, validated by cross-referencing with MALDI-TOF MS and its ability to uncover specific, predictive biomarkers in diseases like colorectal cancer, makes it a superior choice for this application. As Nanopore chemistry and analysis pipelines like Emu continue to improve, the barrier to high-resolution, accessible microbiome analysis will further diminish, firmly establishing ONT's role in the future of clinical and translational microbiome research.
The rapid and accurate identification of pathogens is a cornerstone of effective outbreak investigation and infectious disease control. Next-generation sequencing (NGS) technologies have revolutionized this field by moving beyond traditional culture-based methods, offering unprecedented resolution for tracking transmission routes and characterizing microbial threats. Among available platforms, Illumina and Oxford Nanopore Technologies (ONT) have emerged as prominent tools with complementary strengths and limitations. Illumina sequencing is renowned for its high base-level accuracy, generating millions of short reads with error rates below 0.1% [7]. This technology has become a reference standard for applications requiring precise single-nucleotide variant detection. In contrast, ONT sequencing measures changes in ionic current as single-stranded DNA or RNA passes through a protein nanopore, enabling real-time data analysis and the generation of long reads that can span entire genes or operons [11].
The selection between these platforms represents a critical decision for clinical and public health laboratories, balancing factors such as turnaround time, analytical accuracy, portability, and cost. This guide provides an objective comparison of Illumina and Oxford Nanopore Technologies, focusing on their performance in rapid pathogen detection and outbreak investigation. We present experimental data from recent studies, detailed methodologies, and analytical frameworks to inform platform selection for specific diagnostic and surveillance scenarios in chemogenomic research.
Technology Comparison: Core Characteristics and Performance Metrics
The fundamental differences between Illumina and ONT technologies create distinct performance profiles that influence their suitability for various applications in pathogen detection. Illumina employs sequencing-by-synthesis with fluorescently labeled nucleotides, generating high volumes of short reads (typically 75-300 bp) that provide excellent coverage for variant calling but struggle with repetitive regions and structural variants [7]. ONT's nanopore-based approach directly sequences native DNA or RNA molecules without amplification, producing long reads (commonly 10-100 kb, with ultra-long reads exceeding 100 kb possible) that preserve epigenetic modifications and enable complete assembly of complex genomic regions [11].
Key performance metrics differentiate these platforms, as summarized in Table 1. Historically, ONT has been characterized by higher error rates, though recent improvements in chemistry (R10.4 flow cells) and base-calling algorithms have substantially enhanced accuracy [34] [10]. A 2025 study comparing sequencing quality for Clostridioides difficile genome analysis reported average read qualities of Q25 (99.68% accuracy) for Illumina versus Q15 (96.84% accuracy) for ONT, demonstrating a persistent though narrowing accuracy gap [10].
Table 1: Key Technical Specifications of Illumina and Oxford Nanopore Sequencing Platforms
| Parameter | Illumina | Oxford Nanopore Technologies |
|---|---|---|
| Technology Principle | Sequencing-by-synthesis with reversible dye-terminators | Nanopore-based current measurement |
| Typical Read Length | 75-300 bp (short-read) | 10-100 kb (long-read); up to 100+ kb possible |
| Maximum Output (per instrument) | ~20 Tb (NovaSeq X Plus) | ~28 Tb (PromethION 48) |
| Error Rate | ~0.1% (Q30) | ~1-5% (Q20-Q15); dependent on chemistry |
| Primary Error Mode | Substitution errors | Insertion-deletion errors |
| Time to First Results | Hours to days (after run completion) | Minutes to hours (real-time streaming) |
| Portability | Benchtop to large-scale systems; limited portability | Pocket-sized (MinION) to high-throughput systems |
| Native DNA/RNA Sequencing | No (cDNA only for RNA) | Yes (direct RNA and DNA sequencing) |
| Epigenetic Modification Detection | Requires specialized protocols | Built-in capability (e.g., methylation) |
| Cost per Sample (varies by scale) | Lower for high-throughput projects | Competitive for low-to-medium throughput; decreasing |
ONT's distinctive advantage lies in its real-time data streaming capability, which enables adaptive sampling—a bioinformatics-driven enrichment strategy where sequences of interest are selectively retained while irrelevant DNA is ejected from pores during sequencing [57]. This functionality allows researchers to enrich for pathogen genomes in complex samples without additional laboratory preparation, significantly accelerating time-critical analyses during outbreak investigations.
Experimental Data: Comparative Performance in Pathogen Detection
Bloodstream Infection Diagnostics
Rapid identification of pathogens and antimicrobial resistance (AMR) genes directly from blood cultures is crucial for sepsis management. A 2025 study evaluated ONT's performance for this application using 67 positive blood cultures, limiting sequencing time to just one hour to assess rapid diagnostic potential [58]. The methodology involved collecting positive blood cultures (BACT/ALERT SA bottles), extracting DNA, preparing libraries with ONT rapid barcoding kits, and sequencing on MinION devices with a strict one-hour time limit. Comparative analyses were performed against standard methods: MALDI-TOF-MS for pathogen identification, Illumina NGS for comprehensive AMR gene detection, and antimicrobial susceptibility testing (AST) for resistance phenotyping.
The results demonstrated that ONT sequencing achieved 100% concordance with both MALDI-TOF-MS and Illumina NGS in pathogen identification, detecting all 67 pathogens with 100% sensitivity [58]. For AMR gene detection, ONT identified 584 genes compared to 585 detected by Illumina, yielding a 91.79% consistency rate (537/585 genes). When predicting resistance phenotypes using AST as the reference standard, ONT exhibited competitive performance with Illumina: sensitivity (77.4% vs. 77.3%), specificity (83.3% vs. 83.7%), and accuracy (80.4% for both) [58]. Most notably, the turnaround time for ONT sequencing was 3.5 hours on average, dramatically shorter than Illumina NGS (50.5 hours) and conventional MALDI-TOF-MS plus AST (66-96 hours) [58].
Respiratory Microbiome Profiling
The characterization of respiratory microbial communities presents distinct challenges due to sample complexity and potential host DNA contamination. A 2025 study compared Illumina NextSeq (targeting V3-V4 hypervariable regions) and ONT MinION (full-length 16S rRNA gene) for profiling respiratory samples from ventilator-associated pneumonia patients and a swine model [5]. The experimental protocol involved parallel processing of 34 respiratory samples, with DNA extraction using the Sputum DNA Isolation Kit, followed by platform-specific library preparations: QIAseq 16S/ITS Region Panel for Illumina and ONT 16S Barcoding Kit for MinION sequencing.
The findings revealed platform-specific biases in taxonomic profiling. Illumina captured greater species richness, particularly for low-abundance taxa, while ONT provided superior resolution for dominant bacterial species due to its full-length 16S rRNA coverage [5]. Differential abundance analysis (ANCOM-BC2) indicated that ONT overrepresented certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides). Beta diversity differences were more pronounced in complex porcine samples than in human samples, suggesting that platform effects vary with microbial community complexity [5].
Table 2: Performance Comparison in Clinical Diagnostic Studies
| Study Application | Metric | Illumina Performance | Oxford Nanopore Performance |
|---|---|---|---|
| Bloodstream Infection [58] | Pathogen detection concordance | Reference standard | 100% vs. MALDI-TOF-MS and Illumina |
| AMR gene detection rate | 585 genes detected | 584 genes detected (91.79% concordance) | |
| AST prediction accuracy | 80.4% | 80.4% | |
| Turnaround time | 50.5 hours | 3.5 hours | |
| Respiratory Microbiome [5] | Species richness | Higher | Lower for rare taxa |
| Species-level resolution | Limited (~47%) | Improved (~76%) | |
| Community evenness | Comparable | Comparable | |
| Platform bias | Underrepresents some taxa | Overrepresents Enterococcus, Klebsiella | |
| Gut Microbiome [59] | Species-level classification | 47% | 76% |
| Genus-level classification | 80% | 91% | |
| Read length | 442 ± 5 bp | 1,412 ± 69 bp | |
| Data volume per sample | 30,184 ± 1,146 reads | 630,029 ± 92,449 reads |
Genomic Epidemiology and Outbreak Investigation
For tracking transmission pathways during outbreaks, sequencing platforms must accurately identify genetic relationships between isolates. A 2025 study evaluating C. difficile surveillance found that Illumina data provided superior resolution for epidemiological investigations due to lower error rates [10]. In this study, 37 isolates were sequenced on both platforms, with Illumina libraries prepared using Nextera XT Kits and ONT libraries with rapid barcoding kits (SQK-RBK110-96 and SQK-RBK114-96) on MinION devices using R9.4.1 and R10.4.1 flow cells.
The analysis revealed that Illumina sequencing produced reads with an average quality of 99.68% (Q25), while ONT reached 96.84% (Q15) [10]. This accuracy difference impacted downstream applications: ONT assemblies alone failed to detect certain sequence types (ST5, ST7, ST8, ST13, ST49) that were identified using Illumina. Core genome MLST analysis based on ONT data incorrectly assigned over 180 alleles due to an average of 640 base errors per genome, resulting in less accurate phylogenetic trees for investigating transmission events [10]. However, both platforms performed comparably in detecting key virulence genes (tcdA, tcdB, cdtAB) and tcdC deletions, suggesting ONT remains valuable for virulence profiling when rapid results are prioritized over high-resolution phylogenetics.
Experimental Design and Methodologies
Workflow for Rapid Pathogen Detection from Blood Cultures
The following diagram illustrates the optimized workflow for rapid pathogen identification and AMR gene detection directly from positive blood cultures, as validated in recent studies:
Diagram 1: Rapid pathogen detection workflow using Oxford Nanopore sequencing. The complete process from sample to answer can be completed in 3.5-6 hours, significantly faster than conventional methods.
Adaptive Sampling for Targeted Enrichment
ONT's adaptive sampling functionality enables bioinformatics-based enrichment during sequencing, which is particularly valuable for detecting low-abundance pathogens in complex samples. The following diagram illustrates this process:
Diagram 2: Adaptive sampling workflow for target enrichment. This computational enrichment strategy eliminates the need for physical target enrichment during sample preparation.
Essential Research Reagents and Materials
Successful implementation of rapid pathogen detection protocols requires specific reagents and equipment. Table 3 details essential components for ONT-based diagnostic workflows:
Table 3: Essential Research Reagents and Materials for Rapid Pathogen Detection
| Item | Function | Example Products/Models |
|---|---|---|
| DNA Extraction Kit | Isolation of high-quality genomic DNA from clinical samples | Qiagen EZ1 DNA Tissue Kit, DNeasy PowerSoil Pro Kit [10] |
| Blood Culture System | Automated detection of microbial growth in blood samples | BACT/ALERT SA Aerobic/Anaerobic bottles [58] |
| ONT Library Prep Kit | Preparation of sequencing libraries with barcoding | SQK-RBK114-96 Rapid Barcoding Kit [10] |
| ONT Flow Cells | Nanopore array for sequencing | MinION R10.4.1 flow cells [10] |
| Sequencing Device | Platform for running sequencing experiments | MinION, GridION, PromethION [11] |
| QC Instruments | Quality assessment of nucleic acids | Nanodrop 2000, Qubit 4 Fluorometer [5] |
| Bioinformatic Tools | Data analysis, basecalling, and pathogen identification | Dorado basecaller, EPI2ME Labs, MinKNOW [5] |
Advanced Applications and Future Directions
Emerging Innovations in Clinical Diagnostics
Recent advancements in ONT technology have expanded its applications in clinical diagnostics beyond conventional pathogen detection. A 2025 study demonstrated the use of raw nanopore "squiggle" data combined with artificial intelligence to distinguish viable from dead microorganisms—addressing a key limitation of traditional metagenomic approaches that cannot differentiate DNA from live versus dead cells [60]. This fully computational framework utilizes a Residual Neural Network (ResNet1) to predict microbial viability with high accuracy, potentially transforming infection risk assessment and antibiotic response evaluation without additional laboratory work [60].
In cancer diagnostics, ONT sequencing has enabled dramatically accelerated classification of acute leukemia subtypes. The MARLIN (methylation- and AI-guided rapid leukemia subtype inference) method combines nanopore sequencing with machine learning to achieve 96.2% concordance with conventional diagnostics while reducing classification time from weeks to under two hours [60]. This approach not only matches standard diagnostic accuracy but also identifies cryptic genetic drivers often missed by traditional methods, demonstrating the potential for comprehensive cancer characterization in clinically relevant timeframes.
Environmental Surveillance and One Health Applications
ONT's portability and real-time capabilities make it particularly valuable for environmental surveillance within a One Health framework. A 2025 study monitoring wetland ecosystems utilized passive water samplers combined with nanopore sequencing to profile microbiomes and detect clinically relevant antimicrobial resistance genes [60]. Researchers found that wetlands altered by human activity had over 13-fold more pathogen-associated reads than natural wetlands, demonstrating how this approach can provide insights into the relationships between environmental change, wildlife, livestock, and human health [60].
The same study successfully linked AMR genes to their microbial hosts and tracked avian influenza virus spread, showcasing the technology's capability for integrated pathogen surveillance across ecosystems. This cost-efficient, real-time approach enables early warnings of pathogen and AMR transmission dynamics at the interface of human, animal, and environmental health [60].
The comparative analysis presented in this guide demonstrates that both Illumina and Oxford Nanopore Technologies offer distinct advantages for pathogen detection and outbreak investigation. Illumina remains the preferred platform for applications demanding the highest base-level accuracy, such as high-resolution phylogenetic analysis for precise transmission tracking [10]. Its lower error rates and established bioinformatic pipelines make it ideal for comprehensive genomic epidemiology studies where single-nucleotide precision is essential.
Oxford Nanopore Technologies excels in scenarios requiring rapid turnaround times, portability, and long-read capabilities. ONT's real-time sequencing functionality enables pathogen identification in as little as 1-3 hours of sequencing time [58], making it invaluable for time-critical clinical decisions and field deployment during outbreaks. The technology's ability to generate complete, contiguous assemblies of complex genomic regions provides superior resolution of antimicrobial resistance determinants and mobile genetic elements [34], while its direct RNA sequencing and epigenetic modification detection offer unique insights into gene expression and regulation in pathogens.
For clinical laboratories and public health agencies, strategic platform selection should be guided by specific application requirements rather than seeking a universal solution. A hybrid approach—using ONT for rapid initial assessment and Illumina for confirmatory, high-resolution analysis—may offer an optimal balance of speed and precision for comprehensive outbreak investigation. As nanopore sequencing accuracy continues to improve with advancements in chemistry and base-calling algorithms, ONT's role in clinical diagnostics and public health surveillance is likely to expand, potentially enabling truly comprehensive pathogen characterization in near-real-time during future infectious disease emergencies.
In the field of chemogenomics research, where understanding the genetic basis of drug response and resistance is paramount, the choice of sequencing technology profoundly impacts data quality and biological insights. Illumina and Oxford Nanopore Technologies (ONT) have emerged as dominant sequencing platforms, each with distinct performance characteristics that create a technological trade-off. Illumina sequencing delivers exceptional accuracy (≥99.9%, typically Q30 or above) through its short-read, high-throughput approach, making it ideal for single nucleotide variant calling and quantitative applications [61]. In contrast, ONT generates long reads (thousands to tens of thousands of bases) that span repetitive regions and structural variants but with a higher error rate (recently improved to approximately Q26, or 99.75% accuracy with Dorado basecalling) [61]. This fundamental dichotomy has spurred the development of hybrid sequencing strategies that synergistically combine Illumina's base-level accuracy with Nanopore's long-range information.
Hybrid sequencing represents a paradigm shift for complex genomic analyses in chemogenomics, enabling researchers to overcome limitations inherent to either platform used independently. By integrating these technologies, scientists can achieve highly contiguous and accurate genome assemblies, comprehensively identify structural variations, and resolve complex genomic regions that remain inaccessible to short-read technologies alone [62]. This approach is particularly valuable for studying antimicrobial resistance mechanisms, cancer genomics, and host-pathogen interactions—all central themes in drug discovery and development. The following sections provide a detailed comparison of both platforms, experimental data supporting their complementary nature, and practical methodologies for implementing hybrid sequencing in chemogenomics research.
Technology Comparison: Illumina vs. Oxford Nanopore
The performance characteristics of Illumina and Oxford Nanopore sequencing technologies differ significantly across multiple parameters that directly impact their utility for chemogenomics applications. The table below summarizes these key differences based on current experimental evidence:
Table 1: Performance comparison between Illumina and Oxford Nanopore sequencing platforms
| Parameter | Illumina | Oxford Nanopore |
|---|---|---|
| Read Length | Short (50-300 bp) [62] | Long (5,000-100,000+ bp) [62] |
| Accuracy | High (≥99.9%, typically Q30+) [61] | Moderate (99.75% with Dorado basecalling, ~Q26) [61] |
| Error Profile | Low error rate, occasional amplification biases and issues with GC-rich regions [61] | Higher error rate, struggles with homopolymeric regions and indel errors [61] [22] |
| Typical Applications | Variant calling, RNA-seq, population studies, broad microbial surveys [61] [62] | Structural variation, isoform detection, de novo assembly, species-level resolution [5] [62] |
| Throughput | Very high (NovaSeq X Plus: up to 16 Tb per dual run) [61] | Moderate to high [62] |
| Cost per Base | Low [62] | Higher [62] |
| Time to Results | Fast (whole human genome in <30 hours) [61] | Very fast (real-time analysis, whole genome in ~2 hours) [61] |
| Portability | Benchtop systems available, but generally not portable | High (MinION is pocket-sized and portable) [61] |
The quantitative performance differences between these platforms have been demonstrated across multiple studies. In respiratory microbiome research, Illumina captured greater species richness while ONT provided improved resolution for dominant bacterial species [5]. For genomic characterization of Clostridioides difficile, Illumina sequencing produced reads with an average quality of 99.68% (Q25), while Nanopore sequencing produced reads reaching an average quality of 96.84% (Q15), showing a tenfold difference in quality [22]. Similarly, in tracking aquatic invasive species, Illumina sequencing remained more efficient than Nanopore for detecting species from environmental DNA samples, with Nanopore being less effective at assigning reads at a species level [63].
Experimental Evidence: Comparative Performance Studies
Microbial Genomics and Pathogen Surveillance
Multiple recent studies have directly compared Illumina and Nanopore performance for bacterial genome characterization, providing valuable insights for chemogenomics researchers. In a 2025 study on Streptococcus pneumoniae characterization, both technologies successfully identified the bacterium, enabled serotyping, antimicrobial resistance (AMR) profiling, and GPSC prediction. However, the newer ONT V14 chemistry with R10.4.1 flow cells significantly improved both MLST and pbp prediction in long-read sequencing compared to previous versions. The study concluded that hybrid assembly produced circular and contiguous genomes with high N50 parameters, with long-read assembly followed by short-read polishing being a fast and reliable approach when ONT sequencing depth exceeded 100× [27].
A comprehensive analysis of respiratory microbiomes revealed platform-specific biases in taxonomic profiling. ANCOM-BC2 differential abundance analysis highlighted that ONT overrepresented certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides). The study found that alpha and beta diversity metrics differed between platforms, with Illumina capturing greater species richness, though community evenness remained comparable. Notably, beta diversity differences were significant in pig samples but not in human samples, suggesting that sequencing platform effects are more pronounced in complex microbiomes [5].
Genome Assembly and Variant Calling
The hybrid sequencing approach demonstrates particular utility for genome assembly and structural variant detection, as evidenced by several studies:
Table 2: Performance metrics for different sequencing approaches in genome assembly
| Approach | Assembly Contiguity | Variant Calling Accuracy | Structural Variant Detection | Error Rate |
|---|---|---|---|---|
| Short-Read Only | Fragmented assemblies, gaps likely [62] | High for SNPs and small indels [62] | Poor, often misses large variants [62] | Very low [62] |
| Long-Read Only | Near-complete, fewer gaps [62] | Moderate, struggles with homopolymers [22] | Excellent, spans breakpoints [62] | Higher, ~0.015% substitution rate [22] |
| Hybrid Approach | Highly contiguous and accurate assemblies [62] | High for all variant types [62] [64] | Comprehensive [62] | Very low (after correction) [62] |
In a Clostridioides difficile study, Nanopore sequences exhibited an average of 640 base errors per genome (~0.015% substitution rate), which was reflected by the incorrect assignment of over 180 alleles in core genome multilocus sequence typing (cgMLST) analysis. As a result, Nanopore-derived phylogenies were not as accurate as the Illumina reference, making them inadequate for precise investigation of transmission events. However, both platforms provided comparable, satisfactory results for detecting virulence genes tcdA, tcdB, cdtAB and in-frame deletions in tcdC [22]. The study concluded that while Nanopore has a higher error rate that limits application for high-resolution epidemiological surveillance, its short analysis time, lower cost, and simpler procedure make it valuable when fast, less detailed analyses are preferred.
Methodological Implementation: Hybrid Sequencing Workflows
Experimental Design and Sequencing Protocols
Implementing successful hybrid sequencing requires careful experimental design and platform-specific protocols. For Illumina sequencing, typical library preparation involves fragmenting DNA, attaching adapters, and PCR amplification. For example, in respiratory microbiome studies, the V3-V4 hypervariable region of the 16S rRNA gene is amplified using specific primers (e.g., QIAseq 16S/ITS Region Panel) with the following amplification program: denaturation at 95°C for 5 min; 20 cycles of denaturation at 95°C for 30 s; primer annealing at 60°C for 30 s, extension at 72°C for 30 s; and final elongation at 72°C for 5 min [5].
For Nanopore sequencing, library preparation utilizes different kits, such as the ONT 16S Barcoding Kit (e.g., SQK-16S114.24). Barcoded libraries are pooled and loaded onto a MinION flow cell (e.g., R10.4.1), with sequencing performed using MinKNOW software (e.g., v24.02.16) until the end of the flow cell's life (typically 72 hours) [5]. Recent advancements in flow cell chemistry, such as the R10.4.1 flow cells with Kit14 chemistry (ONT_V14), have significantly improved performance for applications like MLST and pbp prediction in bacterial sequencing [27].
Bioinformatics Pipelines for Hybrid Data Integration
The true power of hybrid sequencing emerges during bioinformatic integration of the data. Several robust pipelines have been developed specifically for this purpose:
Hybrid Assembly Workflow: After initial quality control (using tools like FastQC for Illumina data and NanoPlot for Nanopore data), hybrid assembly can be performed using assemblers like Unicycler or SPAdes with hybrid mode enabled. For bacterial genomes, a recommended approach involves long-read assembly followed by short-read polishing, which is fast and reliable when ONT sequencing depth exceeds 100× [27]. For lower coverage (<50×), tools that perform short-read-first assembly, such as Unicycler, are recommended [27].
Viral Reconnaissance Pipeline: The nf-core/viralrecon pipeline provides a robust framework for analyzing viral samples using both Illumina and Nanopore data. For Illumina short reads, the pipeline performs read QC (FastQC), adapter trimming (fastp), read alignment (Bowtie 2), primer sequence removal (iVar for amplicon data), and variant calling with multiple callers (iVar variants, BCFTools). For Nanopore data, it utilizes the ARTIC Network's field bioinformatics pipeline for alignments, variant calling, and consensus sequence generation [65].
Metagenomic Analysis: Tools like the Arcadia-Science/metagenomics Nextflow workflow enable QC, evaluation, and profiling of metagenomic samples using both short- and long-read technologies [66]. These pipelines are particularly valuable for chemogenomics studies investigating complex microbial communities or host-microbe interactions in response to compound treatment.
Essential Research Reagents and Tools
Successful implementation of hybrid sequencing strategies requires specific laboratory reagents and bioinformatics tools. The following table details key resources mentioned in experimental protocols across the cited studies:
Table 3: Essential research reagents and tools for hybrid sequencing experiments
| Category | Item | Specific Example | Function/Application |
|---|---|---|---|
| Library Preparation | DNA Extraction Kit | Sputum DNA Isolation Kit (Norgen Biotek) [5] | High-quality DNA extraction from complex samples |
| Illumina Library Prep | QIAseq 16S/ITS Region Panel (Qiagen) [5] | Target amplification and library preparation for Illumina | |
| Nanopore Library Prep | ONT 16S Barcoding Kit SQK-16S114.24 [5] | Barcoding and library preparation for Nanopore | |
| Sequencing | Illumina Platform | NextSeq Sequencing Platform [5] | Short-read sequencing with high accuracy |
| Nanopore Platform | MinION Mk1C with R10.4.1 flow cell [5] | Portable long-read sequencing | |
| Bioinformatics | Quality Control | FastQC, NanoPlot, MultiQC [5] [65] | Quality assessment of raw sequencing data |
| Read Processing | Cutadapt, DADA2, Dorado basecaller [5] | Adapter trimming, error correction, basecalling | |
| Hybrid Assembly | Unicycler, SPAdes [22] [27] | Genome assembly using both short and long reads | |
| Variant Calling | iVar, BCFTools [65] | Identification of genetic variants | |
| Taxonomic Classification | Silva 138.1 prokaryotic SSU database [5] | Microbiome profiling and taxonomic assignment |
Hybrid sequencing strategies effectively leverage the complementary strengths of Illumina and Nanopore technologies to overcome the limitations of either platform used independently. The integration of Illumina's high accuracy with Nanopore's long-range information enables comprehensive genomic analyses that are particularly valuable for chemogenomics applications, including antimicrobial resistance profiling, structural variant detection in cancer genomics, and complex microbiome studies. As both technologies continue to evolve—with Illumina enhancing its long-range capabilities through linked-read technologies and Nanopore steadily improving its basecalling accuracy—the relative advantages and implementation details of hybrid approaches will likewise advance.
Future developments in hybrid sequencing will likely focus on streamlining workflows, reducing turnaround time, and improving analytical pipelines for integrated data analysis. The recent introduction of simplified hybrid capture approaches that eliminate bead-based capture and post-hybridization PCR already demonstrates significant potential for improving efficiency and variant calling accuracy [64]. For chemogenomics researchers, these advancements will provide increasingly powerful tools for unraveling the genetic determinants of drug response, resistance mechanisms, and host-pathogen interactions, ultimately accelerating drug discovery and development pipelines.
Optimizing Data Quality: Addressing Error Rates, Bioinformatics, and Experimental Design
In chemogenomic applications research, where precise genetic data informs drug discovery and mechanism-of-action studies, the choice of sequencing platform and corresponding bioinformatics pipeline is paramount. The fundamental differences in chemistry between Illumina (short-read, sequencing-by-synthesis) and Oxford Nanopore Technologies (ONT) (long-read, electronic signal-based) sequencing generate distinct error profiles that must be managed with specialized computational tools. For Illumina data, the DADA2 pipeline represents a gold-standard for amplicon analysis, modeling and correcting its characteristic substitution errors. In contrast, the higher raw error rates and different error spectrum of Nanopore data necessitate custom, often sample-specific, pipelines that leverage consensus strategies and newer chemistries to achieve accuracy. This guide objectively compares the performance of these approaches, providing the experimental data and protocols necessary for researchers to make informed decisions that ensure data fidelity in critical chemogenomic research.
Fundamental Error Profiles: A Tale of Two Technologies
The inherent error profiles of Illumina and Nanopore technologies stem from their distinct sequencing chemistries, necessitating different bioinformatics correction philosophies.
Illumina (Short-Read): This technology is characterized by very low raw error rates (often <0.1%), with errors primarily consisting of substitutions (incorrect base calls). These errors arise during the sequencing-by-synthesis cycles and are not random; their probability increases sharply as sequencing quality declines along the read length [67]. This predictable nature allows for model-based error correction within tools like DADA2, which learns the specific error rates of each transition (e.g., A→C) and uses this model to denoise amplicon sequencing data, resolving true biological sequences (Amplicon Sequence Variants, ASVs) that differ by as little as one nucleotide [68].
Oxford Nanopore (Long-Read): ONT data has a historically higher raw error rate (typically 4-15%), though recent chemistry improvements (R10.4.1) have pushed accuracy above 99% [69] [5] [70]. The error profile is dominated by insertions and deletions (indels), particularly within homopolymeric regions (stretches of the same base) and other low-complexity sequences. This is because the electronic signal from such regions is more challenging for basecalling algorithms to interpret consistently [71] [70]. The error rate is also influenced by sequence context and GC content, with high-GC reads exhibiting more errors [70].
Table 1: Fundamental Characteristics of Illumina and Oxford Nanopore Sequencing
| Feature | Illumina | Oxford Nanopore Technologies (ONT) |
|---|---|---|
| Read Length | Short (up to ~300 bp, 2x250 bp common) | Long (routinely >10 kb, up to several Mb) |
| Primary Error Type | Substitutions | Insertions and Deletions (Indels) |
| Typical Raw Error Rate | < 0.1% [72] | 4% - 15% (Highly dependent on chemistry and basecaller) [69] [5] [70] |
| Key Error Causes | Phasing/pre-phasing during synthesis cycles | Signal interpretation in homopolymers and low-complexity regions [70] |
| Inherent GC Bias | Low | Moderate to High (Error rate higher in high-GC sequences) [70] |
Experimental Protocols for Error-Managed Sequencing
To ensure reliable results, specific wet-lab and computational protocols must be followed for each platform. The following workflows detail the standard methods for 16S rRNA amplicon sequencing, a common application in microbiome-focused chemogenomics.
DADA2 Pipeline for Illumina Amplicon Data
The DADA2 workflow (version 1.16 or later) is a reference-free method that processes demultiplexed Illumina fastq files into an ASV table.
Wet-Lab Protocol (Illumina 16S Amplicon):
- Library Preparation: Amplify the V3-V4 hypervariable region of the 16S rRNA gene using primers (e.g., 341F/806R) [5].
- Sequencing: Sequence on an Illumina MiSeq or NextSeq platform to generate paired-end reads (e.g., 2x300 bp) [67] [5].
Bioinformatics Protocol (DADA2 in R):
- Filter & Trim: Remove low-quality sequences based on expected errors (
maxEE), truncate reads at positions where quality drops (e.g.,truncLen=c(240,160)for forward/reverse reads), and remove phiX contamination [67]. - Learn Error Rates: Estimate the error model from the data itself.
- Sample Inference (Denoising): Apply the error model to distinguish true biological sequences from errors, inferring exact ASVs [68].
- Merge Paired Reads: Combine forward and reverse reads to create the full-length amplicon sequence.
- Construct ASV Table: Create a frequency table of sequences across samples and remove chimeras.
The following diagram illustrates the core DADA2 workflow for Illumina data.
Figure 1: The DADA2 denoising workflow for Illumina amplicon data.
Custom Pipelines for Nanopore Amplicon Data
No single pipeline is as universally established for Nanopore as DADA2 is for Illumina. Successful analysis often involves multi-step custom pipelines focused on generating high-accuracy consensus sequences from raw, error-prone long reads.
Wet-Lab Protocol (Nanopore Full-Length 16S Amplicon):
- Library Preparation: Amplify the full-length 16S rRNA gene (~1,500 bp) and prepare the library using the ONT 16S Barcoding Kit (e.g., SQK-16S114) [5].
- Sequencing: Load the library onto a MinION or PromethION flow cell (preferably R10.4.1 or later) and sequence for 12-72 hours using MinKNOW software [5].
Bioinformatics Protocol (Custom Consensus-Based Pipeline):
- Basecalling & Demultiplexing: Convert raw signal (fast5) to sequence (fastq) and assign samples by barcode using the
doradobasecaller with a high-accuracy (HAC) or super-accuracy (SUP) model [69] [5]. - Read Filtering: Remove low-quality reads and short sequences.
- Clustering & Consensus Building: This is the critical error-correction step.
- Tools like
amplicon_sorterorNGSpeciesIDcluster reads by similarity (e.g., by unique molecular identifier UMI or gene identity) [69]. - Within each cluster, a high-accuracy consensus sequence is built, effectively "averaging out" the random errors present in individual reads.
- Tools like
- Taxonomic Classification: Assign taxonomy to the final consensus sequences using a reference database like Silva 138.1 [5].
The PRONAME pipeline is an example of a dedicated, user-friendly workflow that implements such strategies, integrating "Nanopore-specific quality filtering, clustering and error correction" to produce consensus sequences with ≥99.5% accuracy [73]. Another study achieved highly accurate, "Illumina-like" consensus barcodes that were "generally indel-free" using amplicon_sorter on R10.3 data [69].
Performance Comparison in Microbial Community Profiling
Direct comparisons in 16S rRNA profiling studies highlight the trade-offs between the two platforms and their respective data processing methods. A 2025 study comparing Illumina NextSeq and ONT MinION for respiratory microbiome analysis provides key quantitative insights [5].
Table 2: Performance Comparison in 16S rRNA Profiling of Respiratory Samples [5]
| Performance Metric | Illumina NextSeq (V3-V4 region) | ONT MinION (Full-length 16S) |
|---|---|---|
| Target Region & Read Length | ~300 bp (V3-V4 hypervariable region) | ~1,500 bp (Full-length 16S gene) |
| Taxonomic Resolution | Reliable genus-level classification | Enables species-level and strain-level resolution |
| Species Richness (Alpha Diversity) | Captured greater species richness | Captured lower richness compared to Illumina |
| Community Evenness (Alpha Diversity) | Comparable to Nanopore | Comparable to Illumina |
| Differential Abundance Bias | Detected a broader range of taxa; overrepresentation of Prevotella, Bacteroides | Overrepresentation of Enterococcus, Klebsiella |
| Key Strengths | High accuracy, superior for broad microbial surveys | Long reads provide high taxonomic resolution, portability, real-time data |
The data shows that Illumina with DADA2 remains superior for detecting rare taxa and capturing absolute richness, a consequence of its ultra-high sequencing depth and accuracy. However, Nanopore's long reads provide a clear advantage in taxonomic resolution, enabling confident characterization at the species level.
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table lists key reagents and materials required to perform the benchmark experiments described in this guide.
Table 3: Essential Research Reagents and Materials for Platform Comparison
| Item | Function / Description | Example Product / Kit |
|---|---|---|
| 16S rRNA Gene Primer Set (V3-V4) | Amplifies the target hypervariable region for Illumina short-read sequencing. | 341F/806R primers [5] |
| 16S rRNA Gene Primer Set (Full-length) | Amplifies the entire ~1,500 bp 16S gene for Nanopore long-read sequencing. | Included in ONT 16S Barcoding Kit |
| Illumina Library Prep Kit | Prepares amplicon libraries for sequencing on Illumina platforms. | QIAseq 16S/ITS Region Panel (Qiagen) [5] |
| Nanopore Library Prep Kit | Prepares amplicon libraries for sequencing on ONT platforms. | ONT 16S Barcoding Kit 24 V14 (SQK-16S114.24) [5] |
| ONT Flow Cell | The consumable containing nanopores for sequencing. | MinION Flow Cell (R10.4.1) [5] |
| Reference Database | Curated collection of reference sequences for taxonomic classification. | Silva 138.1 prokaryotic SSU database [5] |
The choice between Illumina/DADA2 and Nanopore/custom pipelines is not a matter of which is universally better, but which is optimal for the specific goals of a chemogenomic research project.
Select Illumina with the DADA2 pipeline when: The primary objective is the sensitive discovery of microbial composition and the detection of low-abundance taxa in a complex community. This is typical in exploratory studies linking community shifts to compound treatment. Its high accuracy and maturity make it the most reliable choice for large-scale, quantitative studies where reproducibility and depth are critical [5] [68].
Select Nanopore with a custom consensus pipeline when: The research question requires high taxonomic resolution down to the species or strain level, or the identification of specific sequence variants (e.g., single-nucleotide polymorphisms) within a gene of interest. This is crucial when tracing strain-level responses to chemical treatments or aiming for functional insights based on precise genotype. Its portability and real-time capability also make it ideal for rapid, in-field diagnostics [69] [5].
For the most comprehensive results, a hybrid approach can be considered, using Illumina for deep, quantitative community profiling and Nanopore for fully resolving the genetic context of key taxa identified. By understanding and strategically managing the distinct error profiles of each platform, chemogenomic researchers can generate the most reliable and informative data to drive drug discovery and development.
Selecting the optimal bioinformatics pipeline is a critical step in next-generation sequencing (NGS) analysis, directly impacting the accuracy and reliability of research outcomes. This guide provides an objective comparison of assemblers and classifiers for Illumina and Oxford Nanopore Technologies (ONT) platforms, drawing on recent experimental data to inform their use in chemogenomic applications.
Sequencing Platforms at a Glance: Illumina vs. Oxford Nanopore
The choice of sequencing platform dictates the available bioinformatic strategies. The table below summarizes the core characteristics of the two leading technologies.
Table 1: Fundamental Comparison of Illumina and Oxford Nanopore Sequencing Technologies
| Feature | Illumina | Oxford Nanopore (ONT) |
|---|---|---|
| Core Technology | Sequencing-by-synthesis (SBS) with reversible dye-terminators [74] | Nanopore-based electronic signal measurement [75] |
| Read Length | Short reads (up to ~2x300bp); Synthetic Long Reads (ICLR) ~6-7 kb [76] | Long reads (can exceed 1 Mb); capable of full chromosome reads [75] |
| Typical Raw Read Accuracy | Very high (Q30+: >99.9% accuracy) [74] | Lower than Illumina (Q15-Q20: 96-99% accuracy); improving with new chemistries and models (up to Q26 simplex) [77] [75] |
| Key Strengths | High accuracy for variant calling, established gold standard for epidemiology [77] [78] | Long reads resolve complex regions, real-time analysis, detect base modifications [75] |
| Primary Limitations | Short reads struggle with repeats and structural variants [76] | Higher error rate can hinder high-resolution phylogenetic analysis [77] [78] |
Assembly Strategies and Performance
Genome assembly is the process of reconstructing a complete genome from sequencing reads. The performance of different assembly strategies varies significantly between platforms.
Assembly of Short-Read (Illumina) Data
For viral genomes from Illumina short-read data, the choice of reference-based assembler is crucial. A 2024 benchmark study of four open-source pipelines using HIV-1 data revealed key performance differences [79].
Table 2: Performance Comparison of Viral Genome Assemblers for Illumina Short-Read Data
| Pipeline | Performance with Matched Reference | Performance with Divergent Reference | Runtime & Resource Profile |
|---|---|---|---|
| Shiver / DShiver | High quality (genome fraction, mismatch/indel rates) [79] | Robust performance with non-matching subtypes [79] | Longer runtime; Dockerized version (DShiver) offers ease of use [79] |
| SmaltAlign | High quality [79] | Robust performance with non-matching subtypes [79] | Fast runtime; user-friendly [79] |
| Viral-ngs | High quality [79] | Performance declines with divergent samples [79] | Fast runtime; lower computational resource requirements [79] |
| V-Pipe | High quality [79] | Performance declines with divergent samples [79] | Longer runtime; provides the broadest functionalities for variant calling [79] |
Assembly of Long-Read (ONT) Data and Hybrid Strategies
While ONT long reads excel at resolving genomic structures, their higher error rate presents a challenge for assemblers. A 2025 study on pathogenic bacteria found that assembly quality varies across species and tool combinations, and errors can persist even in final assemblies, sometimes affecting core genome MLST (cgMLST) loci [78]. Long-read polishing (using the long-read data itself iteratively) generally improves quality, but one round is typically sufficient, as further rounds may degrade assembly [78]. Hybrid assembly (polishing ONT assemblies with high-accuracy Illumina reads) remains a gold standard for maximizing accuracy, but may not be feasible for all labs [78].
A new entrant is the Illumina Complete Long Read (ICLR) assay, which synthetically generates long reads from short-read data. A 2025 metagenomics study found ICLR assemblies had contiguity comparable to ONT assemblies but with higher base-level accuracy and more complete draft genomes [76].
Diagram 1: Bioinformatics assembly strategies and workflows for different sequencing platforms.
Classification and Taxonomic Profiling
In metabarcoding studies (e.g., 16S rRNA for bacteria, ITS for fungi), the bioinformatics pipeline for differentiating taxa significantly influences the resulting community profile.
- OTU vs. ASV Methods: A 2024 comparison of fungal ITS data analysis found that the mothur pipeline (clustering reads into Operational Taxonomic Units, OTUs, at 97% similarity) produced more homogeneous results across technical replicates and detected higher richness than DADA2 (inferring Amplicon Sequence Variants, ASVs). DADA2's high-resolution ASVs may overinflate fungal diversity due to intragenomic variation in the ITS region [80].
- Platform-Specific Pipelines: For 16S rRNA sequencing, the optimal pipeline often depends on the platform. While DADA2 is standard for Illumina and PacBio HiFi data, its denoising algorithm is less suitable for ONT data due to the technology's different error profile. ONT 16S data often requires specialized pipelines like Spaghetti that use OTU-clustering approaches [59].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Sequencing and Analysis Workflows
| Item | Function | Example Use Case |
|---|---|---|
| TruSeq SBS Chemistry | High-accuracy sequencing-by-synthesis reagents for Illumina platforms [74]. | Provides the foundation for high-fidelity short-read data, crucial for variant calling and polishing. |
| Nanopore 6B4 Polishing Chemistry & APK | Reagents designed to improve consensus accuracy for ONT sequencing, part of the Telomere-to-Telomere (T2T) bundle [75]. | Enables highly accurate (Q50) nanopore-only genome assemblies, reducing reliance on Illumina polishing. |
| DNeasy PowerSoil Kit | Standardized DNA extraction kit for complex samples like soil and feces [59]. | Ensures high-quality, inhibitor-free genomic DNA is obtained for downstream amplification and sequencing. |
| 16S Barcoding Kit (SQK-16S024) | ONT kit for amplifying and barcoding the full-length 16S rRNA gene [59]. | Allows for multiplexed, long-read amplicon sequencing for microbial community profiling. |
| SILVA Database | Curated database of aligned ribosomal RNA sequences [59]. | Serves as a reference for taxonomic classification of 16S rRNA amplicon sequences across platforms. |
The selection of a bioinformatics pipeline is not one-size-fits-all and must be aligned with the sequencing platform and research goals.
- For maximum accuracy and high-resolution epidemiology: Illumina sequencing paired with a reference-based assembler like SmaltAlign (for speed) or Shiver (for handling diversity) is the most robust choice. This is critical for detecting single-nucleotide variants or for working with low-diversity pathogens [77] [79].
- For structural genomics and resolving complex regions: ONT sequencing is superior. To mitigate its higher error rate, use latest basecalling models and polishing strategies, ideally with Illumina short-read hybrid polishing for the highest accuracy in final assemblies [75] [78]. The new Illumina Complete Long Read (ICLR) technology presents a promising alternative, offering long-range information with high native accuracy [76].
- For metabarcoding studies: The choice between OTU-clustering (mothur) and ASV-calling (DADA2) should be informed by the target marker. For fungal ITS, OTU clustering is currently more reliable, while DADA2 is well-established for 16S rRNA Illumina data. For ONT amplicon data, use specialized pipelines like Spaghetti [80] [59].
Ultimately, the best pipeline is one that is validated for a specific application. Researchers should leverage controlled experiments or mock communities to benchmark their chosen bioinformatics tools, ensuring their data supports confident scientific conclusions in chemogenomic research and drug development.
In chemogenomic applications, from antimicrobial drug discovery to understanding compound-gene interactions, the accuracy of sequencing data is paramount. Next-generation sequencing (NGS) technologies, primarily Illumina and Oxford Nanopore Technologies (ONT), have become indispensable tools in this field. However, the biological conclusions drawn from their data are inherently risked by technical biases introduced during library preparation and sequencing [81]. These biases, particularly those related to guanine-cytosine (GC) content, primer selection, and DNA extraction methods, can systematically distort representation of biological material, leading to inaccurate genomic reconstructions or misleading microbial abundance profiles in drug treatment studies. For researchers engaged in high-stakes drug development, a precise understanding of these biases is not merely academic; it is a fundamental prerequisite for valid experimental outcomes. This guide provides an objective, data-driven comparison of Illumina and Nanopore platforms, focusing on the sources and mitigation of these critical technical biases to inform robust experimental design in chemogenomic research.
Technical biases in sequencing workflows can compromise data integrity by altering the expected representation of nucleotides, genes, or species. The most significant sources are GC content, library preparation chemistry, and primer selection.
GC Content: DNA molecules with extremely high or low GC content are notoriously challenging to sequence accurately. GC bias manifests as uneven coverage, where sequences with "optimal" GC content (e.g., 45-65%) are over-represented, while GC-rich and GC-poor sequences are under-represented. This can create coverage gaps in genome assemblies and skew quantitative abundance estimates in metagenomic surveys [81].
Library Preparation Chemistry: The enzymatic steps in library preparation are a major source of bias. PCR amplification is a well-known contributor, as it inefficiently amplifies GC-rich templates [81]. Furthermore, transposase-based "rapid" kits (used by both platforms) exhibit sequence-specific insertion preferences. For example, the MuA transposase in ONT rapid kits has a recognized motif (5’-TATGA-3’), leading to biased interaction frequencies and uneven coverage [82]. Ligation-based kits generally provide more uniform coverage but can still under-represent sequences with extreme terminal nucleotide compositions [82].
Primer Selection (16S rRNA Amplicon Sequencing): In amplicon-based microbiome studies, the choice of primers targeting hypervariable regions of the 16S rRNA gene directly influences taxonomic profiling. Full-length 16S sequencing (enabled by long reads) offers higher taxonomic resolution, while short-read sequencing of specific regions (e.g., V3-V4) may struggle with species-level discrimination [5]. PCR conditions during library prep can also introduce bias; for instance, increasing denaturation time has been shown to improve the representation of GC-rich species [83].
Platform-Specific Bias Profiles: An Objective Comparison
Illumina Platform Biases
Illumina's short-read sequencing-by-synthesis is known for high per-base accuracy but is susceptible to specific biases rooted in its library preparation and flow-cell chemistry.
GC Coverage Bias: Illumina workflows, particularly those involving PCR, exhibit major GC biases. A comprehensive 2020 study found that MiSeq and NextSeq workflows were "hindered by major GC biases," with problems becoming "increasingly severe outside the 45–65% GC range." Genomic windows with 30% GC content had over 10-fold less coverage than windows near 50% GC [81]. This coverage bias directly threatens the accuracy of quantitative applications like metagenomic abundance estimates and copy number variant calling.
Primer-Induced Bias in RNA-Seq: A specific bias in transcriptome sequencing is caused by random hexamer priming during cDNA synthesis. This induces a distinct, reproducible bias in the nucleotide composition at the very beginning of sequencing reads, affecting the uniformity of read distribution along transcripts [84]. This bias is independent of the sequencing platform itself but is a consequence of the standard Illumina RNA-Seq library prep protocol.
16S Amplicon Bias: The accuracy of 16S rRNA gene sequencing on Illumina platforms is influenced by genomic GC content. A study on the Ion Torrent PGM platform (a similar semiconductor technology) found that the measured relative abundance of species correlated negatively with their genomic GC content. This suggests a PCR bias against GC-rich species during library preparation, which can be partially mitigated by optimizing PCR conditions [83].
Table 1: Key Bias Characteristics of the Illumina Platform
| Bias Type | Underlying Cause | Impact on Data | Supporting Experimental Data |
|---|---|---|---|
| GC Coverage Bias | PCR amplification during library prep [81] | >10-fold coverage drop for 30% GC regions vs. 50% GC regions; under-representation of extreme GC sequences [81] | Sequencing of 14 bacterial isolates with GC content from 28.9% to 62.4% [81] |
| RNA-Seq Priming Bias | Random hexamer priming during cDNA synthesis [84] | Non-uniform distribution of reads along expressed transcripts; skewed positional coverage [84] | Analysis of nucleotide frequencies at read starts across multiple RNA-Seq experiments [84] |
| 16S Amplicon Bias | PCR bias against high-GC templates; short read length [83] [5] | Underestimation of GC-rich Proteobacteria; overestimation of GC-poor Firmicutes; limited species-level resolution [83] [5] | Sequencing of a 20-member equimolar bacterial mock community [83] |
Oxford Nanopore Platform Biases
Oxford Nanopore Technology (ONT) utilizes a fundamentally different approach, measuring changes in electrical current as DNA strands pass through a protein nanopore. Its long-read capability and lack of PCR in many workflows alter its bias profile.
GC Content and Sequencing Errors: While ONT is often noted for lacking GC bias in coverage [81], GC content remains a crucial parameter for error rate. A 2021 study found that low-GC reads had fewer errors (∼6%) than high-GC reads (∼8%). Approximately half of all sequencing errors occur in homopolymeric regions or regions with short repeats, predominantly manifesting as deletions [85].
Library Prep-Driven Coverage Bias: The choice of ONT library kit significantly influences coverage uniformity. Ligation-based kits (e.g., SQK-LSK109) show relatively even coverage distribution across varying GC contents. In contrast, transposase-based rapid kits (e.g., SQK-RBK110.96) exhibit a strong interaction bias, with enriched cleavage in AT-rich regions (30-40% GC) and a sharp drop in coverage for regions with 40-70% GC content. This bias directly translates to uneven sequencing depth across the genome [82].
Homopolymer Resolution: A historical challenge for ONT has been accurately resolving homopolymer tracts (stretches of identical nucleotides). This can lead to indels in the consensus sequence, though recent improvements in basecalling algorithms and pore chemistry (R10.4.1) have enhanced performance [22] [85].
Table 2: Key Bias Characteristics of the Oxford Nanopore Platform
| Bias Type | Underlying Cause | Impact on Data | Supporting Experimental Data |
|---|---|---|---|
| GC-Error Rate Bias | Signal base-calling complexity in high-GC regions [85] | ~2% higher error rate in high-GC reads vs. low-GC reads; errors often deletions in homopolymers [85] | Benchmarking on 12 bacterial and 2 human datasets sequenced on R9.4.1 flow cells [85] |
| Library Kit Coverage Bias | Transposase (MuA) insertion preference in rapid kits [82] | Normalized coverage decline from +0.07x at 39% GC to -0.11x at 51% GC (rapid kit); ligation kit is more even [82] | Analysis of bovine ear tissue and rumen microbiome DNA comparing ligation vs. rapid kits [82] |
| Homopolymer Indels | Challenges in interpreting current signal across identical nucleotides [85] | Incorrect allele calls adjacent to homopolymers; lower consensus accuracy in repetitive regions [86] [22] | Multiplex amplicon sequencing of 30 human loci; a variant adjacent to a 5-nucleotide homopolymer was not resolved [86] |
| Minimal GC Coverage Bias | PCR-free sequencing; direct detection of native DNA [81] | No significant under-coverage in high or low GC regions; "not afflicted by GC bias" in coverage [81] | Same study of 14 bacterial isolates with a wide range of GC contents [81] |
Experimental Data and Methodologies for Bias Assessment
To ensure the reproducibility of bias assessments, this section outlines key experimental protocols used in the cited studies.
Protocol: Assessing GC Bias Across Sequencing Workflows
This methodology is adapted from a 2020 study that systematically evaluated GC bias across multiple platforms [81].
- Sample Selection: Isolate genomic DNA from a diverse panel of microorganisms with a wide range of known GC contents. The referenced study used 14 different bacteria with mean GC contents ranging from 28.9% to 62.4%.
- Library Preparation and Sequencing: Prepare sequencing libraries from each isolate using standardized, platform-specific protocols (e.g., Illumina Nextera, ONT ligation sequencing). Sequence all libraries to a sufficient depth (>50x).
- Bioinformatic Analysis:
- Read Mapping: Trim raw reads for quality and adapter content. Map reads to the corresponding reference genomes.
- Coverage Calculation: Divide each reference genome into non-overlapping windows (e.g., 1 kb). Calculate the mean read coverage for each window.
- GC Correlation: Calculate the GC content for each genomic window. Plot the normalized coverage (e.g., coverage in window / mean genome coverage) against the GC content of the window.
- Expected Output: The result is a GC-coverage curve. An unbiased workflow will show a flat profile, while a biased one will show a curve, typically peaking in the 45-65% GC range and dropping off at the extremes [81].
Protocol: Evaluating 16S rRNA PCR Bias with a Mock Community
This method, derived from Laursen et al. (2017), uses a defined control to quantify amplification bias [83].
- Mock Community: Utilize a well-defined, validated, and equimolar bacterial mock community, such as the "Microbial Mock Community B" from BEI Resources, which contains 20 bacterial species with known 16S rRNA gene copy numbers and genomic GC content.
- Library Preparation: Perform 16S rRNA gene amplification (e.g., of the V3 region) using your standard primers and PCR conditions. It is critical to use non-degenerative universal primers that perfectly match all mock community sequences to isolate bias to the PCR process itself.
- Sequencing and Data Analysis: Sequence the amplicons on your chosen platform. Process reads through a standard amplicon analysis pipeline (quality filtering, OTU clustering, taxonomy assignment).
- Bias Quantification: Compare the observed relative abundances of each species to the expected even distribution (e.g., 5% for each of 20 species). Calculate the log2(observed/expected) ratio. A strong negative correlation between this ratio and the genomic GC content of the species indicates a significant PCR bias against GC-rich templates [83].
Comparative Workflow Diagram
The following diagram illustrates the key steps in the two major library preparation methods for Oxford Nanopore sequencing, highlighting where biases are introduced.
The Scientist's Toolkit: Key Reagents and Solutions
Table 3: Essential Research Reagents for Mitigating Sequencing Bias
| Reagent / Kit | Function | Role in Bias Mitigation |
|---|---|---|
| PCR-Free Library Prep Kits (e.g., Illumina TruSeq DNA PCR-Free) | Prepares sequencing libraries without PCR amplification. | Eliminates PCR-induced GC bias, providing more uniform coverage across regions with extreme GC content [81]. |
| High-Fidelity DNA Polymerases (e.g., KAPA2G Robust) | Amplifies target DNA with high accuracy in PCR. | Reduces error rates and can improve amplification efficiency of difficult templates, mitigating some PCR bias [86]. |
| Defined Mock Communities (e.g., BEI Resources HM-276D) | Contains genomic DNA from known species in equimolar ratios. | Serves as a process control to quantify and correct for technical bias (GC, amplification) within a specific lab protocol [83]. |
| ONT Ligation Sequencing Kits (e.g., SQK-LSK109) | Prepares DNA libraries using ligation-based, PCR-free methods. | Provides more uniform coverage compared to transposase-based kits, minimizing sequence-specific coverage biases [82]. |
| PCR Additives (e.g., Betaine, DMSO) | Alters DNA melting temperature and polymerase fidelity. | Can improve amplification of GC-rich regions (betaine) or reduce secondary structures, helping to balance representation [81]. |
The choice between Illumina and Nanopore for chemogenomic research is not a matter of selecting a universally superior technology, but of aligning platform strengths with specific research objectives while actively managing their respective biases.
- For applications demanding the highest single-base accuracy (e.g., variant calling for resistance mutations, SNP discovery), Illumina remains the gold standard [22] [4]. However, researchers must account for its significant GC coverage bias through the use of PCR-free library prep and careful interpretation of data from extreme GC regions [81].
- For applications requiring long-range genomic context (e.g., resolving complex resistance loci, phage integration sites, eukaryotic genomes), Nanopore is unparalleled [4]. Its key advantage is the absence of GC coverage bias in PCR-free workflows [81]. The primary challenge is a higher raw error rate, particularly in homopolymers and high-GC regions, necessitating sufficient sequencing depth for accurate consensus [85]. To ensure even coverage, ligation-based kits are strongly recommended over rapid transposase-based kits for quantitative applications [82].
A forward-looking strategy involves hybrid sequencing approaches, leveraging Illumina's accuracy to polish Nanopore's long reads, thus combining comprehensive genomic context with high-fidelity base calling [22] [4]. Regardless of the platform, the consistent use of internal controls like mock communities is essential for benchmarking and validating the performance of any chosen workflow, ensuring that biological conclusions are built upon a foundation of technically robust data [83].
In next-generation sequencing (NGS) workflows, library preparation is not merely a preliminary step but a critical determinant of overall success. It is estimated that over 50% of sequencing failures or suboptimal runs can be traced back to issues encountered during library preparation [87]. The process of converting RNA or DNA samples into a format compatible with sequencing platforms involves multiple delicate steps where inefficiencies can introduce artifacts, biases, and quantitative inaccuracies that compromise data quality. For chemogenomic applications research—where researchers investigate how chemicals affect cellular networks—maintaining library integrity is particularly crucial for detecting true biological signals against background noise.
The choice between Illumina and Oxford Nanopore Technologies (ONT) platforms introduces distinct considerations for library preparation. While both require the attachment of platform-specific adapters to DNA fragments, their underlying chemistries, error profiles, and optimal workflows differ substantially [88] [89]. This guide objectively compares library preparation best practices for these leading platforms, with particular focus on avoiding adapter artifacts and ensuring library quality—factors that directly impact the reliability of chemogenomic insights.
Platform Comparison: Key Technical Characteristics
Table 1: Technical comparison of Illumina and Oxford Nanopore library preparation and sequencing characteristics.
| Characteristic | Illumina | Oxford Nanopore Technologies |
|---|---|---|
| Sequencing Chemistry | Sequencing by synthesis with reversible terminators | Nanopore-based current disruption measurement |
| Read Length | Short reads (typically 100-600 bp) [90] | Long reads (typically >10 kb) [88] |
| Adapter Ligation Approach | Ligation of duplex adapters via T-A cloning [91] | Ligation-based or rapid transposase-based chemistry [88] |
| Typical Input Requirements | 1-5 μg for mechanical shearing; less for enzymatic [91] | 1 μg for long fragments; 100-200 fmol for short fragments [88] |
| Primary Error Mode | Substitution errors [10] | Insertion-deletion errors, particularly in homopolymers [92] [10] |
| PCR Requirement | Often required for amplification of adapter-ligated fragments [87] | Optional; PCR-free protocols available [88] |
| Raw Read Accuracy | >99.9% (Q30) [90] | ~96-99% (Q15-Q20+) depending on chemistry [59] [10] |
Library Preparation Methodologies: A Step-by-Step Comparison
Core Workflow Stages
Both Illumina and Nanopore library preparation share several fundamental steps, though implementation details differ significantly.
Fragmentation: DNA must be fragmented into appropriate sizes for sequencing. Mechanical shearing (sonication, nebulization, or focused acoustics) offers minimal sequence bias but requires specialized equipment. Enzymatic fragmentation uses nucleases or transposases (tagmentation) and is amenable to automation with lower input requirements, though it may introduce sequence-specific biases [87] [91]. For Illumina, the typical fragment size ranges from 300-600 bp, while Nanopore routinely handles fragments >10 kb [88] [91].
End Repair and A-Tailing: After fragmentation, DNA ends are converted to a uniform format for adapter ligation. This involves blunting (filling in or removing overhangs), 5' phosphorylation, and adding a single 3' adenosine overhang for T-A cloning with Illumina adapters [87] [91]. Nanopore ligation-based kits similarly require end repair to create ligation-compatible ends [88].
Adapter Ligation: Platform-specific adapters containing necessary functional elements (primer binding sites, barcodes) are ligated to fragments. Illumina uses duplex adapters ligated to both ends of fragments [91], while Nanopore offers both ligation-based and rapid transposase-based adapter attachment, with the latter combining fragmentation and adapter incorporation in a single step [88]. Proper adapter stoichiometry is critical to minimize adapter-dimer formation in both systems [87].
Library Amplification (Optional): PCR amplification may be required for low-input samples or specific protocols. However, excessive amplification can introduce biases and duplicate reads. Nanopore more readily supports PCR-free sequencing, potentially preserving base modifications and reducing bias [88].
Quality Control and Quantification: Accurate library quantification is essential for optimal sequencing performance. qPCR-based methods are recommended for Illumina as they selectively quantify full-length library fragments, unlike fluorometric methods that may overestimate concentration by including adapter dimers and incomplete products [93]. Nanopore libraries are typically quantified using fluorometric methods (e.g., Qubit) with consideration of fragment length for molarity calculations [88].
Workflow Visualization
Library Preparation Workflow Comparison: This diagram illustrates the key steps in library preparation for Illumina and Oxford Nanopore Technologies platforms, highlighting points where methodological differences emerge.
Experimental Data: Performance Comparison in Microbial Applications
Taxonomic Resolution in 16S rRNA Sequencing
A 2025 comparative study of rabbit gut microbiota analyzed identical samples across Illumina (MiSeq), PacBio (HiFi), and Oxford Nanopore (MinION) platforms, providing direct performance comparisons for amplicon sequencing [59].
Table 2: Taxonomic classification resolution across sequencing platforms for 16S rRNA gene sequencing [59].
| Taxonomic Level | Illumina MiSeq | PacBio HiFi | ONT MinION |
|---|---|---|---|
| Phylum Level | >99% | >99% | >99% |
| Family Level | >99% | >99% | >99% |
| Genus Level | 80% | 85% | 91% |
| Species Level | 47% | 63% | 76% |
The study demonstrated that while all platforms performed comparably at higher taxonomic ranks, Nanopore provided superior species-level resolution, classifying 29% more sequences to species level compared to Illumina [59]. However, a significant limitation across all platforms was that most species-level classifications were assigned ambiguous names such as "uncultured_bacterium," highlighting database limitations rather than technological constraints [59].
Error Profiles and Their Implications
Error characteristics differ substantially between platforms, influencing appropriate applications and necessary quality control measures.
Illumina exhibits low error rates (<0.1%) dominated by substitution errors, making it suitable for applications requiring high base-level accuracy such as single nucleotide variant detection [90] [10]. A 2025 study on Clostridioides difficile surveillance reported Illumina read quality of Q25 (99.68% accuracy) versus Nanopore's Q15 (96.84% accuracy) [10].
Nanopore has higher overall error rates (~4-8% raw read error) with a predominance of insertion-deletion errors, particularly in homopolymer regions and repetitive sequences [92] [10]. However, the recent R10.4.1 flow cells with Kit 14 chemistry have improved accuracy to over 99% (Q20+) [88]. Error profiles are also highly reproducible and systematic rather than random, enabling correction through bioinformatic approaches [92].
These differences significantly impact analytical outcomes. In the C. difficile study, Nanopore sequences exhibited approximately 640 base errors per genome compared to Illumina, resulting in incorrect assignment of over 180 alleles in core genome multilocus sequence typing (cgMLST) analysis and rendering Nanopore-derived phylogenies inadequate for high-resolution epidemiological surveillance [10].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key research reagents and materials for library preparation and quality control.
| Reagent/Material | Function | Platform Application |
|---|---|---|
| DNeasy PowerSoil Pro Kit (Qiagen) | DNA extraction from complex matrices | Both platforms [59] [10] |
| Covaris AFA systems | Focused acoustic shearing for DNA fragmentation | Primarily Illumina [87] [91] |
| Nextera XT DNA Library Prep Kit (Illumina) | Tagmentation-based library preparation | Illumina [59] [10] |
| Ligation Sequencing Kit V14 (ONT) | Ligation-based library preparation | Nanopore (optimized for R10.4.1) [88] |
| Rapid Barcoding Kit (ONT) | Rapid library prep with integrated barcoding | Nanopore [10] |
| AMPure XP beads (Beckman Coulter) | Size selection and cleanup of libraries | Both platforms [59] [90] |
| KAPA Library Quantification Kit (Roche) | qPCR-based accurate library quantification | Primarily Illumina [93] |
| Qubit dsDNA HS Assay Kit (Thermo Fisher) | Fluorometric DNA quantification | Both platforms [88] [90] |
| Agilent Bioanalyzer/Fragment Analyzer | Size distribution assessment | Both platforms [59] [88] |
| CAPTORs (Control Adaptors) | Internal reference controls for accuracy monitoring | Primarily Nanopore [92] |
Quality Control Strategies: Preventing and Detecting Adapter Artifacts
Adapter Artifact Prevention
Adapter artifacts—including adapter-dimer formation, incomplete adapter ligation, and off-target adapter hybridization—can significantly compromise sequencing data quality and yield. Prevention strategies include:
Optimized Adapter Stoichiometry: Using the correct adapter-to-insert ratio is critical. Excess adapters increase dimer formation, while insufficient adapters reduce library yield [87]. Illumina recommends qPCR-based quantification which selectively amplifies only full-length adapter-ligated fragments, unlike fluorometric methods that also detect free adapters and dimers [93].
Size Selection: Implementation of rigorous size selection using magnetic beads (e.g., AMPure XP) or gel extraction effectively removes adapter dimers (typically ~120-150 bp) from legitimate library fragments [87]. For Nanopore libraries, size selection can also enrich for desired fragment length ranges.
Library Quantification Best Practices: qPCR-based methods are strongly recommended for Illumina libraries as they specifically quantify fragments containing complete P5 and P7 adapter sequences necessary for cluster formation [93]. Fluorometric methods (e.g., Qubit) risk overestimating functional library concentration by including adapter dimers and partially constructed fragments [93]. UV spectrophotometry should be avoided due to inability to distinguish adapter-ligated fragments from free nucleotides and other contaminants [93].
Quality Control Metrics and Validation
Implementation of robust QC checkpoints throughout library preparation is essential for detecting potential artifacts before sequencing:
Fragment Size Distribution: Automated electrophoresis systems (Bioanalyzer, TapeStation, Fragment Analyzer) verify expected size distributions and detect adapter-dimer contamination [93] [88]. For Illumina libraries with broad size distributions, Bioanalyzer quantification is not recommended due to decreasing accuracy with increasing fragment size distribution [93].
Internal Reference Controls: For Nanopore sequencing, CAPTORs (Control Library Adaptors) can be integrated into library preparation to provide ongoing measurement of sequencing accuracy and quantitative performance [92]. These adaptors contain defined control sequences that enable per-read accuracy assessment and can identify systematic errors.
Mock Communities: Inclusion of synthetic microbial communities with known composition validates quantitative accuracy and detects biases introduced during library preparation [90] [92]. Studies demonstrate that while both platforms correctly identify majority community members, relative abundance estimates can vary significantly between platforms [90].
Platform Selection Guide for Chemogenomic Applications
The choice between Illumina and Nanopore technologies for chemogenomic research depends on specific experimental goals and quality requirements:
Choose Illumina when: Your application requires high single-base accuracy for variant calling [10], you're working with low-input samples where amplification is unavoidable [87], or you need standardized, established protocols for regulatory applications [7].
Choose Nanopore when: Your research benefits from long reads to span repetitive regions or structural variants [88] [10], you need real-time sequencing capabilities for adaptive sampling [10], you want to detect base modifications directly without specialized protocols [88], or portability is important for field applications [7] [10].
For comprehensive chemogenomic studies, a hybrid approach leveraging both technologies can provide the benefits of both: Illumina's accuracy for SNP detection and Nanopore's long reads for resolving complex genomic regions [10]. The increasing accuracy of Nanopore sequencing, particularly with recent chemistry improvements (Kit 14, R10.4.1 flow cells), continues to narrow the performance gap for applications requiring high base-level accuracy [88].
In the field of chemogenomic applications research, the choice between Illumina and Oxford Nanopore Technologies (ONT) sequencing platforms presents a significant strategic decision. Each technology generates data with distinct characteristics—Illumina produces high-volume, accurate short reads, while ONT yields longer reads with higher error rates. These fundamental differences necessitate platform-specific data filtering strategies to optimize taxonomic assignments and minimize false positive classifications, which is particularly crucial in drug development contexts where accurate microbial community profiling can inform therapeutic discovery.
The higher error rates historically associated with ONT sequencing (5-15% for older chemistries) [5] present unique bioinformatic challenges for taxonomic classification, while Illumina's short-read data requires different approaches to overcome its limitations in species-level resolution. This guide systematically compares experimental data from both platforms and provides evidence-based filtering protocols to enhance data quality, with particular emphasis on applications relevant to researchers, scientists, and drug development professionals working in chemogenomics.
Platform Comparison: Technical Specifications and Performance Metrics
Fundamental Technological Differences
Table 1: Core Technology Comparison Between Illumina and Oxford Nanopore Platforms
| Feature | Illumina | Oxford Nanopore Technologies (ONT) |
|---|---|---|
| Sequencing Principle | Sequencing-by-synthesis with reversible dye-terminators [4] | Measurement of current changes as DNA passes through protein nanopores [4] |
| Read Length | Short reads (100-300 bp) [5] [94] | Long reads (hundreds of bases to >4 Mb) [4] |
| Error Mode | Substitution errors [10] | Insertion-deletion errors predominantly [10] |
| Raw Read Accuracy | High (>99.9%) [10] [4] | Variable (96-99% with newer chemistries) [5] [10] |
| Typical Applications | Genus-level profiling, variant calling, high-precision applications [5] [10] | Species-level resolution, structural variant detection, real-time analysis [5] [4] |
Experimental Performance in Taxonomic Classification
Comparative studies reveal how these technological differences translate to practical performance in taxonomic classification tasks:
Table 2: Experimental Performance Metrics for Taxonomic Classification
| Metric | Illumina Performance | Nanopore Performance | Experimental Context |
|---|---|---|---|
| Species-Level Classification Accuracy | Limited due to short read length [5] | Enhanced with full-length 16S rRNA sequencing [5] | 16S rRNA profiling of respiratory communities [5] |
| Error Rate Impact on Classification | Minimal effect on species-level assignment [95] | Filtering shorter reads improves performance despite higher error rates [95] | Benchmarking of 16 common bloodstream pathogens [95] |
| Alpha Diversity Capture | Greater species richness [5] | Comparable community evenness [5] | Respiratory microbiome analysis [5] |
| False Positive Rate | Lower false positives with standard databases [96] | Higher false positives, requiring abundance filtering [96] | Mock community evaluation [96] |
| Best-Performing Classifier | Bracken (standard database): 97.8% correct species assignment [95] | Long-read specific tools (BugSeq, MEGAN-LR) outperform short-read tools [96] | Multiple benchmarking studies [95] [96] |
Data Filtering Strategies and Experimental Protocols
Platform-Specific Quality Control and Filtering
Effective data filtering begins with platform-specific quality control processes. For Illumina data, FastQC provides comprehensive quality assessment through per-base quality plots, sequence duplication levels, and adapter contamination analysis [97]. The typical workflow involves trimming low-quality bases (typically below Q20) and removing adapter sequences using tools like Cutadapt [5]. For Nanopore data, NanoPlot offers specialized quality assessment for long-read data, with filtering based on quality scores and read length [97]. The SeqKit tool can then filter reads based on established quality thresholds [97].
Research indicates that filtering ONT reads shorter than 2 kilobases significantly improves taxonomic classification precision by reducing false positives [96]. This length-based filtering approach compensates for the technology's higher error rate while leveraging its primary advantage of long-range sequence information. For both platforms, removing host DNA contamination is crucial, particularly in clinical samples where human DNA can dominate the sequence data [98].
Taxonomic Classification and Database Selection
The choice of taxonomic classification tool and reference database significantly impacts false positive rates. Studies demonstrate that Bracken with a standard database (30-50 GB) achieves median species-level identification of 97.8% for both Illumina and Nanopore data [95]. However, performance varies substantially by database size, with mini databases (8 GB) yielding significantly poorer results (median 86.4% correct species assignment) [95].
DNA-to-DNA classification methods (e.g., Kraken2, Bracken) generally outperform DNA-to-protein and marker-based methods for both platforms [95]. However, for Nanopore data specifically, tools designed for long reads (e.g., MetaMaps, MEGAN-LR, BugSeq) demonstrate superior performance by utilizing the long-range information across multiple genes [96]. These tools leverage the ability of long reads to span multiple taxonomic marker regions, thereby increasing classification confidence.
Data Filtering Workflows for Illumina and Nanopore Platforms
Advanced Filtering: Abundance Thresholds and Misclassification Catalogues
Sophisticated filtering approaches involve establishing abundance thresholds based on known misclassification patterns. Research shows that taxonomic misclassification is not random but occurs more frequently between species with higher Average Nucleotide Identity (ANI) [95]. By creating species-specific misclassification catalogues, researchers can establish abundance thresholds to distinguish true polymicrobial infections from bioinformatic artefacts.
For example, in a study of common bloodstream pathogens, Escherichia coli proved particularly challenging to classify correctly, with only 56.1-96.0% of reads assigned to the correct species depending on the tool used [95]. Establishing that a secondary species detection below 4% abundance may represent misclassification (rather than true co-infection) helps reduce false positives in clinical diagnostics [95].
The ANCOM-BC2 differential abundance analysis tool has been successfully employed to identify and correct for platform-specific biases, such as ONT's overrepresentation of Enterococcus and Klebsiella and underrepresentation of Prevotella and Bacteroides [5].
Experimental Protocols for Platform Comparison
Comparative Microbiome Analysis Protocol
A standardized protocol for comparing sequencing platforms enables researchers to establish laboratory-specific filtering parameters:
Sample Collection and DNA Extraction:
- Collect respiratory samples (e.g., from ventilator-associated pneumonia patients) and preserve at -80°C [5]
- Extract genomic DNA using the Sputum DNA Isolation Kit (Norgen Biotek) [5]
- Assess DNA quality and concentration using Nanodrop 2000 spectrophotometer and Qubit 4 fluorometer [5]
Library Preparation and Sequencing:
- For Illumina: Prepare V3-V4 16S rRNA libraries using QIAseq 16S/ITS Region Panel with 20 amplification cycles [5]
- For Nanopore: Prepare libraries using ONT 16S Barcoding Kit SQK-16S114.24 [5]
- Sequence Illumina libraries on NextSeq for 2×300 bp reads [5]
- Sequence Nanopore libraries on MinION Mk1C with R10.4.1 flow cell [5]
Bioinformatic Processing:
- Process Illumina data with nf-core/ampliseq pipeline using DADA2 for error correction [5]
- Process Nanopore data with EPI2ME Labs 16S Workflow following basecalling with Dorado [5]
- Apply platform-specific filtering: quality trimming for Illumina, length and quality filtering for Nanopore [5] [96]
- Perform taxonomic classification against Silva 138.1 database for both platforms [5]
Downstream Analysis:
- Calculate alpha and beta diversity metrics using vegan package in R [5]
- Conduct differential abundance analysis with ANCOM-BC2 [5]
- Compare taxonomic composition at genus and species levels [5]
- Evaluate false positive rates using known mock community compositions [96]
Reagent Solutions for Comparative Sequencing Studies
Table 3: Essential Research Reagents and Tools for Sequencing Comparisons
| Reagent/Tool | Function | Application Context |
|---|---|---|
| DNeasy PowerSoil Kit (QIAGEN) | DNA extraction from complex samples | Standardized DNA isolation for microbiome studies [98] |
| QIAseq 16S/ITS Region Panel | Illumina library preparation for 16S rRNA | Amplification of V3-V4 hypervariable regions [5] |
| ONT 16S Barcoding Kit (SQK-16S114.24) | Nanopore library preparation for full-length 16S | Full-length 16S rRNA gene sequencing [5] |
| AMPure XP beads | DNA purification and size selection | Cleanup of shorter DNA fragments after extraction [98] |
| Silva 138.1 prokaryotic SSU | Reference database for taxonomic classification | Curated 16S rRNA database for uniform classification [5] |
| ZymoBIOMICS Microbial Standards | Mock community controls | Validation of classification accuracy and false positive rates [96] |
Discussion: Implications for Chemogenomic Applications
In chemogenomic research, where understanding microbe-drug interactions is paramount, the complementary strengths of Illumina and Nanopore platforms can be leveraged through integrated approaches. Illumina's high accuracy makes it ideal for initial broad microbial surveys detecting subtle community changes in response to compound exposure [5] [4]. Nanopore's long-read capability provides species-level resolution essential for identifying specific bacterial strains encoding biotransformation pathways [5] [94].
The filtering strategies outlined here directly address key challenges in chemogenomics. By reducing false positives through rigorous bioinformatic filtering, researchers can more confidently identify true microbial biomarkers predictive of drug response. Furthermore, Nanopore's real-time sequencing capability enables rapid functional screening of microbial communities against compound libraries, potentially accelerating discovery timelines [98] [4].
Future methodological developments will likely focus on hybrid sequencing approaches that leverage both technologies' strengths—using Illumina data to polish Nanopore assemblies or developing integrated classification algorithms that simultaneously analyze both data types [5] [98]. As sequencing chemistries continue to improve, with Nanopore's Q20 chemistry achieving 99% accuracy [96], the filtering requirements will evolve, but the fundamental principle of platform-specific data processing will remain essential for reliable taxonomic assignments in chemogenomic research.
Platform Selection Framework for Chemogenomic Applications
Optimized data filtering strategies are essential for maximizing the value of both Illumina and Nanopore sequencing technologies in chemogenomic research. Illumina platforms benefit from rigorous quality trimming and database selection to leverage their high accuracy, while Nanopore data requires length filtering and long-read specific classifiers to overcome higher error rates while capitalizing on superior species-level resolution. The implementation of species-specific abundance thresholds based on misclassification patterns further enhances reliable taxonomic assignment. As both technologies continue to evolve, maintaining platform-specific filtering approaches will remain crucial for generating biologically meaningful results in drug discovery and development applications.
Benchmarking Performance: Direct Comparisons for Taxonomic Resolution and Accuracy
The choice of sequencing technology is a foundational decision in microbial genomics, influencing the resolution, accuracy, and application of research findings. Within chemogenomic applications research, where understanding the interaction between chemicals and microbial genomes is paramount, this choice becomes even more critical. This guide provides an objective, data-driven comparison of two dominant sequencing platforms—Illumina and Oxford Nanopore Technologies (ONT). By evaluating their performance across mock community and complex sample studies, we aim to equip researchers, scientists, and drug development professionals with the evidence needed to select the optimal technology for their specific investigative goals.
Performance Comparison: Key Metrics and Quantitative Data
The following tables summarize critical performance metrics derived from recent comparative studies, highlighting the inherent trade-offs between these two technologies.
Table 1: Platform-wide performance characteristics for microbial community analysis.
| Performance Metric | Illumina | Oxford Nanopore Technologies (ONT) |
|---|---|---|
| Typical Read Length | Short-read (~150-300 bp) [5] | Long-read (up to full-length 16S ~1,500 bp or more) [5] |
| Raw Read Accuracy | >99.9% (Q30) [10] | ~96.8% (Q15) to ~99.8% with latest chemistry [10] [27] |
| Error Profile | Substitution errors [10] | Insertion/Deletion errors [10] |
| Ideal Taxonomic Resolution | Genus-level [5] | Species-level and strain-level [5] [27] |
| Sequencing Speed | Hours to days | Minutes to hours (real-time) [10] |
| Portability | Benchtop or large-scale systems | Portable (MinION) to benchtop [10] |
Table 2: Comparative results from specific microbial studies.
| Study / Metric | Illumina Results | Oxford Nanopore Results |
|---|---|---|
| C. difficile Genome Analysis (2025) [10] | ||
| Average Read Quality | 99.68% (Q25) | 96.84% (Q15) |
| cgMLST Allele Mismatches | Reference (0) | >180 per genome |
| Virulence Gene Detection | Satisfactory | Satisfactory and comparable |
| S. pneumoniae Characterization (2025) [27] | ||
| Serotyping & AMR Prediction | Established standard | Successfully achieved |
| MLST Prediction | High accuracy | Improved with V14 chemistry |
| Respiratory Microbiome 16S Profiling (2025) [5] | ||
| Species Richness (Alpha Diversity) | Higher | Comparable community evenness |
| Species-Level Resolution | Limited | Improved for dominant species |
Experimental Protocols in Cited Studies
The quantitative data presented above are generated by specific, reproducible experimental methodologies. Below are the detailed protocols for the key studies cited.
- Sample Collection: A total of 34 respiratory samples were collected, comprising human specimens from ventilator-associated pneumonia (VAP) patients (n=20) and samples from an experimental swine model of VAP (n=14). All samples were stored at -80°C immediately after collection.
- DNA Extraction: Genomic DNA was extracted using the Sputum DNA Isolation Kit (Norgen Biotek). DNA quality and concentration were assessed using a Nanodrop 2000 spectrophotometer and a Qubit 4 fluorometer.
- Library Preparation & Sequencing:
- Illumina: DNA libraries of the V3-V4 hypervariable region of the 16S rRNA gene were prepared using the QIAseq 16S/ITS Region Panel (Qiagen). The pool of DNA products was sequenced on an Illumina NextSeq platform to generate 2x300 bp paired-end reads.
- Nanopore: Sequencing libraries were prepared with the ONT 16S Barcoding Kit (SQK-16S114.24). Barcoded libraries were pooled and loaded onto a MinION flow cell (R10.4.1) and sequenced on a MinION Mk1C device.
- Data Analysis:
- Illumina Data: Processed using the nf-core/ampliseq workflow. Sequences were processed using DADA2 to generate Amplicon Sequence Variants (ASVs).
- Nanopore Data: Raw reads were basecalled and demultiplexed using the Dorado basecaller. Post-sequencing, reads were processed using the EPI2ME Labs 16S Workflow.
- Downstream Analysis: Taxonomic classification for both platforms was performed against the Silva 138.1 prokaryotic SSU database. Diversity and differential abundance analyses were conducted in R using the
phyloseqandANCOMBCpackages.
- Bacterial Isolates: 37 C. difficile isolates of animal and human origin were included.
- DNA Extraction: For most isolates, a pre-lysis step using enzymatic lysis buffer was followed by automated purification with the MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche). For others, mechanical lysis and extraction were performed with the DNeasy PowerSoil Pro Kit (Qiagen).
- Library Preparation & Sequencing:
- Illumina: Libraries were constructed with the Nextera XT Kit and sequenced on an Illumina NextSeq 500 platform with a 2x150 bp kit.
- Nanopore: Libraries were prepared with rapid barcoding kits (SQK-RBK110-96 or SQK-RBK114-96) and sequenced on a MinION device with R9.4.1 or R10.4.1 flow cells.
- Data Analysis:
- Illumina Data: Paired-end reads were trimmed using Trimmomatic v0.39.
- Nanopore Data: Base-calling and demultiplexing of raw FAST5 files were performed using Guppy v5.0.11. Adapters were removed using
qcat. - Assembly & Typing: Illumina-only (SPAdes), Nanopore-only (Flye), and hybrid (Unicycler) assemblies were generated. Typing analyses included cgMLST and virulence gene detection.
Workflow Visualization: Sequencing and Analysis Pipeline
The following diagram illustrates the generalized experimental workflow for a head-to-head comparison study, as implemented in the cited research.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key reagents and solutions for head-to-head sequencing comparisons.
| Item | Function | Example Use Case |
|---|---|---|
| Sputum DNA Isolation Kit (Norgen Biotek) | Extracts genomic DNA from complex, low-biomass respiratory samples. | Respiratory microbiome studies [5]. |
| QIAseq 16S/ITS Region Panel (Qiagen) | Prepares amplicon sequencing libraries for Illumina, targeting specific hypervariable regions. | 16S rRNA gene sequencing on Illumina platforms [5]. |
| ONT 16S Barcoding Kit (Oxford Nanopore) | Prepares barcoded libraries for full-length 16S rRNA gene sequencing on Nanopore devices. | Full-length 16S analysis on MinION [5]. |
| Nextera XT DNA Library Prep Kit (Illumina) | Prepares sequencing libraries for whole-genome shotgun sequencing on Illumina platforms. | Whole-genome sequencing of bacterial isolates [10]. |
| Rapid Barcoding Kit (Oxford Nanopore) | Enables quick library preparation and multiplexing for whole-genome sequencing. | Whole-genome sequencing of bacterial isolates [10]. |
| DNeasy PowerSoil Pro Kit (Qiagen) | Efficiently extracts DNA from complex environmental and difficult-to-lyse bacterial samples. | DNA extraction from C. difficile and other tough microorganisms [10]. |
| Silva SSU rRNA Database | Provides a curated, high-quality reference database for taxonomic classification of 16S sequences. | Unified taxonomic classification for cross-platform comparison [5]. |
For researchers in chemogenomic applications, the choice between Illumina and Oxford Nanopore Technologies (ONT) sequencing platforms involves a fundamental trade-off between sequence accuracy and taxonomic resolution. Illumina short-read sequencing is characterized by high per-base accuracy but is generally limited to genus-level classification. In contrast, ONT long-read sequencing, despite a higher per-base error rate, provides superior species-level resolution by sequencing the full-length 16S rRNA gene. This guide objectively compares their performance using empirical data to inform platform selection for drug development and microbiomics research.
In chemogenomic research, accurately characterizing microbial communities is crucial for understanding drug-microbiome interactions, identifying pathogenic species, and discovering novel therapeutic compounds. Taxonomic classification serves as the foundational step in this process. The 16S ribosomal RNA (rRNA) gene is the standard biomarker used for bacterial identification, containing nine hypervariable regions (V1-V9) that provide taxonomic specificity. The length of the 16S rRNA gene sequence obtained directly determines the taxonomic resolution—the lowest taxonomic rank (e.g., genus or species) to which a sequence can be reliably assigned.
Genus-level resolution is often sufficient for broad ecological surveys, but species-level identification is critical in clinical and pharmaceutical contexts. Many genera contain species with vastly different functional roles and clinical implications; for example, some Escherichia species are commensals while others are pathogens. The ability to differentiate between them directly impacts the accuracy of host-microbiome association studies and the identification of microbial drug targets.
Platform Performance: Quantitative Comparison of Classification Rates
The following table summarizes the key performance metrics for Illumina, ONT, and PacBio (included for context) platforms based on recent empirical studies:
Table 1: Taxonomic Classification Resolution Across Sequencing Platforms
| Sequencing Platform | Read Length (bp) | Target Region | Genus-Level Classification Rate | Species-Level Classification Rate | Key Advantage |
|---|---|---|---|---|---|
| Illumina MiSeq/NextSeq | ~300 bp (paired-end) | V3-V4 | 80% - 94.79% [59] [99] | 47% - 55.23% [59] [99] | High per-base accuracy (Q25-Q30) [77] |
| Oxford Nanopore (ONT) | ~1,500 bp (full-length) | V1-V9 | 91% [59] | 76% [59] | Real-time sequencing, high species-resolution |
| PacBio HiFi | ~1,450 bp (full-length) | V1-V9 | 85% - 95.06% [59] [99] | 63% - 74.14% [59] [99] | High-fidelity long reads (Q27) [99] |
Table 2: Error Profiles and Practical Considerations for Microbial Community Profiling
| Parameter | Illumina | Oxford Nanopore |
|---|---|---|
| Average Per-Base Accuracy | ~99.9% (Q25-Q30) [77] | ~96.84% (Q15) [77] |
| Error Type | Predominantly substitution errors [95] | Higher indels and substitutions [95] |
| Impact on Typing | Reliable for cgMLST and phylogenetic inference [77] | Higher error limits high-resolution epidemiology [77] |
| Best Application in Chemogenomics | Broad microbial surveys, abundance profiling | Species-level resolution, real-time pathogen ID |
Experimental Protocols for Performance Benchmarking
To ensure the comparability of the data presented in the previous section, understanding the underlying experimental methodologies is essential. The following workflows are standardized from recent comparative studies.
Sample Preparation and Sequencing Protocols
A. Illumina Short-Read Protocol (V3-V4) The Illumina protocol typically follows the 16S Metagenomic Sequencing Library Preparation guide:
- PCR Amplification: The hypervariable V3-V4 regions are amplified using primers such as 341F and 805R [5] [99].
- Library Construction: Nextera XT Indexes are attached for multiplexing [59].
- Sequencing: Sequencing is performed on MiSeq or NextSeq platforms to generate 2 × 300 bp paired-end reads [5] [99]. Bioinformatic Processing: Data is processed using pipelines like nf-core/ampliseq or DADA2 within QIIME2 to correct errors, merge paired-end reads, and generate Amplicon Sequence Variants (ASVs) for taxonomic classification [5] [59].
B. ONT Long-Read Protocol (Full-Length) The ONT protocol leverages the 16S Barcoding Kit:
- PCR Amplification: The full-length 16S rRNA gene (~1,500 bp) is amplified using universal primers 27F and 1492R [59].
- Library Preparation & Sequencing: Barcoded libraries are loaded onto MinION flow cells (e.g., R10.4.1) and sequenced on a Mk1C device for up to 72 hours [5]. Bioinformatic Processing:* Basecalling and demultiplexing are performed using MinKNOW/Dorado. Subsequent analysis often uses EPI2ME or Spaghetti (an OTU-based pipeline), as the higher error rate can make DADA2-based denoising less effective [5] [59].
Diagram 1: Experimental workflow for 16S rRNA sequencing
Taxonomic Classification and Analysis Workflow
After generating sequence variants, the taxonomic assignment process is performed, which is hierarchical in nature, moving from higher to lower taxonomic ranks.
Diagram 2: Hierarchical taxonomic classification
- Reference Database: A curated database (e.g., SILVA, Greengenes) provides a taxonomy of known sequences.
- Classifier: A classification algorithm (e.g., Naive Bayes, Kraken2, HFTC) compares query sequences to the reference.
- Hierarchical Assignment: The classifier assigns taxonomy from the top down (Phylum → Class → ... → Species). Illumina's short reads often provide insufficient information for reliable classification beyond genus level, while ONT's long reads contain enough discriminatory information to proceed to species level [100] [101].
Advanced Concepts in Performance Evaluation
Beyond Binary Metrics: The Average Taxonomy Distance (ATD)
Traditional metrics like accuracy can be misleading for taxonomic classification. The Average Taxonomy Distance (ATD) is a more robust metric that quantifies the degree of misclassification error by considering the taxonomic tree [100].
- Calculation: ATD measures the distance between the predicted and actual taxon in the taxonomic hierarchy. A misclassification at a higher rank (e.g., wrong phylum) is penalized more heavily than a misclassification at a lower rank (e.g., wrong species) [100].
- Advantage: ATD provides a more nuanced view of performance, favoring "stable" methods that may not always be perfectly correct but avoid severe misclassifications. This is particularly valuable for evaluating classifiers on complex, real-world samples [100].
Leveraging Species Abundance for Improved Accuracy
The standard assumption in classification is that all species in a reference database are equally likely to be found. However, this is biologically unrealistic. Bespoke taxonomic weights—which incorporate prior knowledge of species abundance in specific environments—can significantly enhance classification accuracy [102].
- Mechanism: A classifier (e.g., q2-clawback) uses environment-specific taxonomic abundance profiles to inform its decisions. When a sequence could match several similar species, the classifier is "biased" toward the one more commonly found in that habitat [102].
- Impact: One study demonstrated that using bespoke weights reduced the species-level classification error rate from 25% to 14%, achieving species-level accuracy comparable to what was previously only possible at the genus level [102].
The Scientist's Toolkit: Essential Research Reagents and Databases
Table 3: Key Reagents, Tools, and Databases for 16S rRNA Sequencing Studies
| Item | Function / Role | Example Products / Software |
|---|---|---|
| DNA Extraction Kit | Isolate high-quality microbial DNA, critical for long-read amplification. | QIAamp DNA Stool Mini Kit, DNeasy PowerSoil Kit [103] [59] |
| 16S Amplification Primers | Target specific variable regions of the 16S rRNA gene for PCR. | 341F/805R (Illumina V3-V4), 27F/1492R (ONT/PacBio full-length) [59] [99] |
| Sequencing Kit | Library preparation for the respective platform. | Illumina: QIAseq 16S/ITS Panel; ONT: 16S Barcoding Kit (SQK-16S114) [5] |
| Bioinformatic Pipelines | Process raw data: quality control, denoising, clustering, taxonomy assignment. | DADA2 (Illumina/PacBio), nf-core/ampliseq, EPI2ME Labs (ONT), Spaghetti (ONT) [5] [59] |
| Taxonomic Reference Database | Curated collection of reference sequences for classifying unknown reads. | SILVA, Greengenes, UNITE (for fungi) [5] [101] |
| Classification Algorithms | Assign taxonomic labels to sequences based on reference databases. | Kraken2, Bracken, q2-feature-classifier, HFTC (for fungi) [95] [101] |
The choice between Illumina and Nanopore for chemogenomic research is not a matter of which platform is universally superior, but which is optimal for the specific research question.
- Select Illumina when your study requires highly accurate quantification of microbial community structure and relative abundances at the genus level. Its high per-base accuracy makes it ideal for large-scale population studies where reproducibility and detection of broad shifts in community composition are the primary goals [5] [77].
- Select Nanopore when species-level identification is critical and some trade-off in per-base accuracy is acceptable. Its ability to sequence the full-length 16S rRNA gene provides superior resolution for differentiating closely related species, identifying specific pathogens, and discovering novel taxa, which is often paramount in clinical diagnostics and targeted therapeutic development [5] [59].
For the highest data quality, a hybrid approach using both technologies is emerging as a powerful strategy, leveraging Illumina's accuracy to polish and validate assemblies generated from Nanopore's long reads [103].
The choice of sequencing platform is a critical methodological decision in microbial ecology, directly influencing the observed diversity and composition of microbial communities. For researchers employing 16S rRNA gene sequencing, Illumina and Oxford Nanopore Technologies (ONT) represent two widely used yet fundamentally different approaches. Illumina provides high-accuracy, short-read sequences typically targeting hypervariable regions, while ONT generates longer, full-length 16S reads with a historically higher error rate but improved taxonomic resolution. This guide objectively compares how these technical differences translate into variations in alpha and beta diversity estimates, providing drug development professionals and scientists with the experimental data necessary to select the appropriate platform for their chemogenomic research.
Table 1: Platform Comparison and Diversity Findings from Key Studies
| Study Focus | Sequencing Platforms Compared | Key Findings on Alpha Diversity | Key Findings on Beta Diversity |
|---|---|---|---|
| Respiratory Microbiomes (Human & Porcine) [5] | Illumina NextSeq (V3-V4) vs. ONT MinION (Full-length 16S) | Illumina captured greater species richness; community evenness was comparable between platforms [5]. | Significant differences in porcine samples; no significant differences in human samples, suggesting platform effects are more pronounced in complex microbiomes [5]. |
| Gut Microbiota (Rabbits) [59] | Illumina MiSeq (V3-V4) vs. ONT MinION (Full-length) vs. PacBio HiFi (Full-length) | Diversity analysis showed significant differences in taxonomic composition between the three platforms [59]. | PCoA based on Bray-Curtis and Jaccard distances showed significant differences between platforms (PERMANOVA) [59]. |
| Gut Microbiome (Pigs) [104] | Illumina MiSeq (V3-V4) vs. ONT MinION (Full-length 16S) | Not explicitly quantified in results summary. | The two platforms showed compatible results, justifying the use of MinION for field applications [104]. |
Experimental Protocols for Comparative Studies
The comparative findings summarized in this guide are derived from rigorous experimental designs. The following protocols detail the methodologies used in the cited studies, providing a blueprint for researchers seeking to replicate or design similar comparisons.
Protocol 1: Respiratory Microbiome Profiling
This protocol was designed to compare platform performance on low-biomass respiratory samples from both human and animal models [5].
- Sample Collection and DNA Extraction: A total of 34 respiratory samples were collected, including human specimens from ventilator-associated pneumonia (VAP) patients (n=20) and samples from an experimental swine model of VAP (n=14). Genomic DNA was extracted in parallel for both platforms using a commercial kit, and its quality and concentration were assessed [5].
- Library Preparation and Sequencing:
- Illumina NextSeq: DNA libraries of the V3-V4 hypervariable region were prepared using the QIAseq 16S/ITS Region Panel. The pool of DNA products was sequenced to generate 2x300bp paired-end reads [5].
- ONT MinION: Sequencing libraries were prepared using the ONT 16S Barcoding Kit, which covers the full-length 16S rRNA gene. Barcoded libraries were pooled and loaded onto a flow cell, and sequencing was performed for up to 72 hours [5].
- Data Processing:
- Illumina Data: Processed using the nf-core/ampliseq workflow, which included primer trimming with Cutadapt, quality filtering, and denoising into Amplicon Sequence Variants (ASVs) using DADA2 [5].
- Nanopore Data: Raw reads were basecalled and demultiplexed using the Dorado basecaller. Post-sequencing, reads were processed using the EPI2ME Labs 16S Workflow for quality control and taxonomic classification [5].
- Downstream Analysis: All analyses were performed in R. Alpha diversity was assessed using Shannon diversity, and beta diversity was evaluated using standard metrics. Differential abundance analysis was performed with ANCOM-BC2 [5].
Protocol 2: Multi-Platform Gut Microbiota Analysis
This protocol compares three sequencing platforms for characterizing rabbit gut microbiota, with a focus on species-level resolution [59].
- Sample Collection and DNA Extraction: Four samples of soft feces were taken from rabbit does. Bacterial genomic DNA was isolated from the frozen fecal samples using the DNeasy PowerSoil kit [59].
- PCR Amplification and DNA Sequencing:
- Illumina MiSeq: The V3-V4 regions were amplified using Illumina's recommended primers following the 16S Metagenomic Sequencing Library Preparation protocol [59].
- PacBio HiFi: The full-length 16S rRNA gene was amplified using universal primers (27F/1492R) tailed with PacBio barcode sequences. Library preparation used the SMRTbell Express Template Prep Kit, and sequencing was performed on a Sequel II system [59].
- ONT MinION: The full-length 16S rRNA gene (V1–V9) was amplified using the 16S Barcoding Kit with primers 27F/1492R. Sequencing was conducted on a MinION device [59].
- Bioinformatic Analyses:
- Illumina and PacBio: Reads were processed using the DADA2 pipeline in R to generate ASVs [59].
- ONT: Due to higher error rates, reads were analyzed using a custom OTU-based pipeline (Spaghetti) instead of DADA2 [59].
- Taxonomic Annotation: Sequences from all platforms were imported into QIIME2. A Naïve Bayes classifier trained on the SILVA database was customized for each platform's specific primers and read lengths [59].
- Diversity Analysis: Alpha and beta diversity analyses were performed in R using the phyloseq package. Beta diversity was evaluated using PCoA based on Bray-Curtis and Jaccard matrices, and statistical significance was assessed with PERMANOVA [59].
Understanding Diversity Metrics
- Alpha Diversity: Measures the species diversity within a single sample. Common metrics include:
- Beta Diversity: Measures the difference in microbial communities between samples [105].
- Bray-Curtis Dissimilarity: A quantitative measure that takes species abundance into account. It is more powerful for detecting subtle clusters [105].
- Jaccard Index: A qualitative measure based on species presence/absence [105].
- Results are often visualized using Principal Coordinates Analysis (PCoA), where samples cluster based on the similarity of their microbial profiles [105].
Platform Selection Workflow
The diagram below outlines the decision-making process for selecting a sequencing platform based on research priorities, derived from the consensus findings of the compared studies.
Research Reagent Solutions
The following table lists key materials and their functions essential for conducting the experimental protocols cited in this comparison.
| Item Name | Function / Application | Relevant Study |
|---|---|---|
| Sputum DNA Isolation Kit (Norgen Biotek) | Genomic DNA extraction from low-biomass respiratory samples. | [5] |
| QIAseq 16S/ITS Region Panel (Qiagen) | Library preparation for Illumina sequencing of the V3-V4 hypervariable regions. | [5] |
| ONT 16S Barcoding Kit (SQK-16S114.24) | Library preparation for full-length 16S rRNA gene sequencing on Nanopore platforms. | [5] |
| DNeasy PowerSoil Pro Kit (Qiagen) | High-quality DNA extraction from complex samples like soil and feces, effective for Gram-positive bacteria. | [59] [10] |
| Nextera XT Index Kit (Illumina) | Preparation of multiplexed amplicon libraries for Illumina sequencing. | [59] [10] |
| Silva 138.1 SSU Database | A curated reference database for taxonomic classification of 16S rRNA gene sequences. | [5] [59] |
The body of evidence demonstrates that the choice between Illumina and Nanopore sequencing has a measurable and sometimes significant impact on microbial diversity estimates. Illumina tends to capture greater taxonomic richness, making it ideal for broad ecological surveys where detecting rare taxa is paramount. In contrast, ONT's primary strength lies in its superior species-level resolution, enabled by full-length 16S reads, and its operational advantages of portability and rapid turnaround time. The observed differences in beta diversity further indicate that data from these two platforms should not be combined directly without caution. The optimal choice is not a matter of which platform is universally better, but which is better suited to the specific research question, sample type, and analytical requirements of the study. Future advancements in bioinformatics, such as error-correction tools for long reads, and hybrid sequencing approaches promise to further leverage the complementary strengths of both technologies.
High-throughput sequencing technologies have revolutionized the study of microbial communities, enabling detailed characterization of microbiomes in health, disease, and various environmental contexts. Among the most widely used platforms, Illumina short-read and Oxford Nanopore Technologies (ONT) long-read sequencing each offer distinct advantages and limitations that significantly impact downstream analyses, particularly differential abundance (DA) testing. DA analysis aims to identify taxa whose abundances differ significantly between experimental conditions, a fundamental task in microbiome research [106].
The selection of sequencing platform introduces substantial technical biases that can confound biological interpretations. Illumina sequencing, with its high per-base accuracy but short read lengths, provides excellent genus-level resolution but struggles with species-level discrimination. In contrast, ONT sequencing generates full-length 16S rRNA reads enabling superior taxonomic resolution, though historically with higher error rates [5]. These technical differences systematically influence which taxa appear differentially abundant in comparative studies, creating platform-specific taxonomic biases that researchers must understand to draw valid biological conclusions.
This review synthesizes recent evidence on how Illumina and ONT sequencing technologies affect DA analysis results, providing researchers with a framework for selecting appropriate platforms and interpreting findings within the context of platform-specific limitations.
Comparative Performance of Sequencing Platforms
Technical Characteristics and Taxonomic Resolution
Direct comparisons of Illumina and ONT platforms reveal fundamental differences in their operational characteristics and resulting data quality. Illumina sequencing consistently delivers higher raw read accuracy (Q25-Q30) compared to ONT (Q15), representing a tenfold difference in error rates [10]. However, ONT's ability to sequence the entire ~1,500 bp 16S rRNA gene provides a crucial advantage for discriminating closely related bacterial species [5].
Table 1: Platform Characteristics and Taxonomic Resolution
| Parameter | Illumina | Oxford Nanopore |
|---|---|---|
| Read Length | Short reads (~300 bp) targeting hypervariable regions (e.g., V3-V4) | Full-length 16S rRNA gene reads (~1,500 bp) spanning V1-V9 |
| Raw Read Accuracy | ~99.9% (Q30) [10] | ~96.8% (Q15) [10] |
| Error Profile | Substitution errors [10] | Higher insertion-deletion errors [10] |
| Species-Level Classification | 47-48% of sequences [5] [59] | 76% of sequences [5] [59] |
| Genus-Level Classification | 80% of sequences [59] | 91% of sequences [59] |
| Optimal Application | Broad microbial surveys, genus-level profiling [5] | Species-level resolution, real-time applications [5] |
The enhanced resolution of ONT translates to practical advantages in taxonomic classification. Studies demonstrate that ONT classifies 91% of sequences to genus level and 76% to species level, compared to 80% and 47% respectively for Illumina [59]. However, a significant limitation affecting both platforms is that many species-level classifications are assigned ambiguous labels such as "uncultured_bacterium," highlighting database limitations rather than technological constraints [59].
Impact on Diversity Metrics and Community Representation
Sequencing platform selection significantly influences both alpha and beta diversity measures, which are fundamental to microbiome study design and interpretation. Analysis of respiratory microbiome samples revealed that Illumina typically captures greater species richness, while community evenness remains comparable between platforms [5]. Beta diversity differences were particularly pronounced in complex microbiomes (porcine samples) compared to human samples, suggesting that platform effects are more substantial in highly diverse communities [5].
Table 2: Diversity Metrics and Community Representation Across Platforms
| Metric | Illumina | Oxford Nanopore | Notes |
|---|---|---|---|
| Alpha Diversity (Richness) | Higher [5] | Lower | Difference more pronounced in complex microbiomes |
| Alpha Diversity (Evenness) | Comparable [5] | Comparable [5] | Consistent across sample types |
| Beta Diversity | Significant differences in complex samples [5] | Significant differences in complex samples [5] | Platform effects minimal in low-complexity communities |
| Detection of Dominant Taxa | Broader range of taxa [5] | Improved resolution for dominant species [5] | Complementary strengths |
| Relative Abundance Correlation | High correlation between platforms for core taxa [59] | High correlation between platforms for core taxa [59] | Disparities for low-abundance and rare taxa |
Taxonomic profiling reveals that each platform has distinct detection biases. Illumina typically detects a broader range of taxa, while ONT exhibits improved resolution for dominant bacterial species [5]. For example, in gut microbiome studies, the family Lachnospiraceae was reported at nearly double the abundance with ONT (51.06%) compared to Illumina (27.84%) [59]. These systematic differences directly impact which taxa are identified as differentially abundant in case-control studies.
Experimental Protocols for Platform Comparison
Sample Processing and Sequencing Workflows
Standardized protocols for cross-platform comparisons involve parallel processing of samples from DNA extraction through bioinformatic analysis. For respiratory microbiome studies, DNA is typically extracted using commercial kits (e.g., Norgen Biotek Sputum DNA Isolation Kit), with quality assessment via Nanodrop and Qubit fluorometry [5]. The same DNA extract is then used for both Illumina and ONT library preparation to eliminate extraction bias.
For Illumina sequencing, the V3-V4 hypervariable regions of the 16S rRNA gene are amplified using targeted primers (e.g., QIAseq 16S/ITS Region Panel) with a two-step PCR protocol: initial denaturation at 95°C for 5 minutes, followed by 20 cycles of denaturation (95°C for 30s), annealing (60°C for 30s), and extension (72°C for 30s), with final elongation at 72°C for 5 minutes [5]. Libraries are sequenced on Illumina NextSeq to generate 2×300 bp paired-end reads.
For ONT sequencing, the full-length 16S rRNA gene is amplified using primers 27F and 1492R with the ONT 16S Barcoding Kit (SQK-16S114.24) [5] [59]. PCR amplification typically uses 40 cycles [59], followed by library preparation and sequencing on MinION devices using R10.4.1 flow cells, with sequencing continuing for up to 72 hours [5].
Bioinformatic Processing and Differential Abundance Analysis
Bioinformatic processing differs substantially between platforms due to their distinct error profiles and read characteristics. Illumina data typically undergoes quality filtering using FastQC, primer trimming with Cutadapt, and denoising via DADA2 to generate amplicon sequence variants (ASVs) [5]. ONT data requires specialized processing pipelines such as EPI2ME Labs 16S Workflow or Spaghetti, with quality control, filtering, and operational taxonomic unit (OTU) clustering [5] [59]. Both platforms use the same reference databases (e.g., SILVA 138.1) for taxonomic classification to enable direct comparison [5].
Differential abundance analysis employs various statistical methods, each with distinct assumptions and performance characteristics. Common approaches include analysis of compositions of microbiomes with bias correction (ANCOM-BC), ALDEx2, and DESeq2 [5] [106]. Studies consistently show that the choice of DA method significantly impacts results, with different tools identifying drastically different sets of significant taxa [106]. For example, when applied to the same datasets, ALDEx2 and ANCOM-BC produce the most consistent results and agree best with the intersect of results from different approaches [106].
Platform-Specific Biases in Taxonomic Abundance
Systematic Biases in Microbial Community Representation
Comparative analyses consistently reveal that Illumina and ONT platforms systematically over- and under-represent specific bacterial taxa. In respiratory microbiome studies, ANCOM-BC2 differential abundance analysis demonstrated that ONT overrepresents certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides) compared to Illumina [5]. These biases appear consistent across sample types, suggesting they derive from platform biochemistry rather than community composition.
The direction and magnitude of taxonomic biases correlate with genomic features, particularly GC content. ONT library preparation methods exhibit distinct sequence-specific biases: ligation-based kits show underrepresentation of AT-rich sequences, while transposase-based (rapid) kits exhibit preference for specific recognition motifs (5'-TATGA-3') and reduced yield in regions with 40-70% GC content [82]. These biases directly impact the apparent abundance of clinically relevant taxa such as Fusobacterium nucleatum (28% GC), which can be underestimated by up to a factor of two in metagenomic analyses without proper correction [107].
Impact on Biological Interpretation
Platform-specific taxonomic biases can substantially alter biological interpretations, particularly when comparing microbial communities between experimental conditions. Methods like GuaCAMOLE have been developed to correct GC-content-dependent biases in metagenomic data, significantly improving abundance estimates for taxa with extreme GC content [107]. Without such corrections, differential abundance analyses may identify platform-specific technical artifacts rather than biologically meaningful differences.
The consistency of platform biases across studies suggests they represent systematic rather than random errors. Research demonstrates that the same DNA samples sequenced on different platforms yield different DA results, with varying sets of taxa identified as statistically significant [5] [59] [106]. This highlights the critical importance of using consistent sequencing platforms within a study and applying appropriate correction methods when comparing results across studies using different technologies.
The Scientist's Toolkit
Essential Research Reagents and Computational Tools
Table 3: Key Research Reagents and Computational Tools for Cross-Platform Comparisons
| Category | Item | Function | Considerations |
|---|---|---|---|
| Wet Lab Reagents | Sputum DNA Isolation Kit (Norgen Biotek) | DNA extraction from respiratory samples | Consistent extraction across samples is critical |
| QIAseq 16S/ITS Region Panel (Qiagen) | Target amplification for Illumina sequencing | Optimized for V3-V4 hypervariable regions | |
| ONT 16S Barcoding Kit (SQK-16S114.24) | Full-length 16S amplification for Nanopore | Enables single-tube library preparation | |
| DNeasy PowerSoil Kit (Qiagen) | DNA extraction from complex samples | Effective for soil, gut, and environmental samples | |
| Computational Tools | nf-core/ampliseq | Illumina 16S data processing | Reproducible, containerized workflow |
| EPI2ME Labs 16S Workflow | ONT 16S data analysis | User-friendly interface for MinION data | |
| Spaghetti | Custom ONT 16S analysis pipeline | OTU-based approach for higher error rate | |
| GuaCAMOLE | GC bias correction | Alignment-free method for metagenomic data | |
| ANCOM-BC2 | Differential abundance testing | Accounts for compositional nature of data | |
| Reference Databases | SILVA 138.1 | Taxonomic classification | Curated 16S database, regularly updated |
The choice between Illumina and ONT sequencing platforms for differential abundance analysis involves trade-offs between taxonomic resolution, accuracy, and practical considerations. Illumina remains the preferred platform for broad microbial surveys requiring high accuracy and deep sequencing, particularly for genus-level analyses. ONT excels in applications requiring species-level resolution, rapid turnaround time, and the ability to resolve complete 16S rRNA sequences [5].
To mitigate platform-specific biases in differential abundance studies, researchers should:
- Maintain platform consistency within a study to enable valid comparative analyses
- Apply appropriate bias correction methods for GC content and library preparation artifacts [107] [82]
- Use multiple differential abundance methods and focus on the consensus findings to ensure robust biological interpretations [106]
- Consider hybrid approaches that leverage both Illumina and ONT sequencing to capitalize on their complementary strengths [5]
As both technologies continue to evolve, with Illumina improving read lengths and ONT enhancing accuracy, the landscape of platform-specific biases will likewise change. Ongoing validation and comparison studies remain essential for understanding how technical factors influence biological interpretations in microbiome research.
Molecular Inversion Probes (MIPs) represent a powerful targeted sequencing technology that combines the scalability to target numerous genomic regions with a cost-effective, single-reaction library preparation process [108]. As next-generation sequencing (NGS) becomes increasingly integral to microbiological fields, including infectious disease diagnostics, the choice of sequencing platform is critical [109]. This case study provides a direct, data-driven comparison of MIP-based pathogen detection on two prominent sequencing platforms: Illumina (short-read) and Oxford Nanopore Technologies (ONT, long-read). The objective is to evaluate their performance characteristics within the context of chemogenomic applications research, enabling scientists to select the most appropriate technology for specific diagnostic and research goals.
Experimental Design and Methodologies
To ensure a meaningful comparison, the following section outlines the core experimental protocols and bioinformatic approaches used in the cited studies.
Molecular Inversion Probe Assay Workflow
The fundamental MIP assay involves a standardized multi-step process for targeted enrichment, though variations exist between standard and improved protocols.
The diagram above illustrates the core MIP workflow. Key methodological details from the cited studies include:
- Probe Design: MIPs are designed with target-specific arms flanking a universal linker sequence. Studies utilized tools like MIPgen for design, aiming for amplicon lengths of 225–300 bp for short-read platforms [110] [108].
- Multiplex MIP Capture: The hybridization mix, containing DNA and the MIP pool, is subjected to a thermal cycling protocol (e.g., 98°C for 3 min, then a gradient down to 56°C for incubation) to facilitate specific probe binding. The gap-filling and ligation step is performed using a enzyme mix containing Ampligase and a DNA polymerase (e.g., Phusion HF or Q5 High-Fidelity), which closes the probe into a circular molecule [108].
- Clean-up: Linear, non-circularized probes and other contaminants are removed through enzymatic digestion with Exonuclease I and III [108].
- Library Preparation: The circularized MIP products are amplified via PCR using primers that add platform-specific sequencing adapters and sample barcodes, ready for sequencing on either Illumina or ONT platforms [109] [108].
Improved MIP (iMIP) Protocol
A significant methodological advancement, termed "improved MIP" (iMIP), was developed to address limitations of standard protocols. Key modifications include [108]:
- Reduced Hybridization Time: The hybridization incubation was shortened from overnight to 60 minutes.
- Enhanced Enzymes: The use of Q5 High-Fidelity DNA Polymerase instead of Phusion HF during gap filling.
- Optimized Clean-up: Doubling the concentration of exonuclease enzymes and reducing the digestion time to 10 minutes.
- This iMIP protocol achieved a turnaround time of approximately 4 hours, significantly improved uniformity, and provided better coverage of GC-rich regions, achieving >95% on-target reads [108].
Direct Performance Comparison: Illumina vs. ONT
A head-to-head study evaluating the same MIP panel on both Illumina and ONT platforms provides the most direct evidence for performance comparison. The data below summarizes key findings from this analysis.
Table 1: Direct Performance Metrics of MIP Sequencing on Illumina vs. ONT Platforms [109]
| Performance Metric | Illumina Platform | Oxford Nanopore Platform |
|---|---|---|
| Genus-level Concordance (31 bacterial pathogens) | 96.7% | 90.3% |
| Viral/Parasitic Detection (18 organisms at 10⁴ PFU/mL) | Detected most targets | Detected most targets (with few exceptions) |
| Read Characteristics | Greater raw read counts, but lower percent mapped reads | Fewer raw read counts |
| Impact on Limit of Detection (LOD) | No significant difference compared to ONT | No significant difference compared to Illumina |
| Predictive Values (vs. qRT-PCR on chikungunya samples) | PPV: 0.91, NPV: 1.00 | PPV: 0.88, NPV: 1.00 |
Analysis of Comparative Data
The data in Table 1 reveals several critical insights for researchers:
- Accuracy: Illumina demonstrated a higher concordance rate for bacterial pathogen identification, which is consistent with its reputation for higher per-base accuracy [109] [10]. A separate study on Clostridioides difficile reported Illumina read quality at Q25 (99.68% accuracy) versus Nanopore at Q15 (96.84% accuracy), a tenfold difference in error rate [10].
- Sensitivity: Both platforms were largely equivalent in detecting low concentrations of viral and parasitic agents, and both achieved perfect negative predictive value (NPV=1.00) in a mock clinical validation [109]. This indicates that MIPs on either platform are highly reliable for ruling out infection when the test is negative.
- Data Utility: While Illumina produced more raw reads, the lower mapping efficiency suggests a higher proportion of off-target or technical artifacts. Despite producing fewer total reads, ONT's data was sufficient to achieve a similar LOD, indicating efficient on-target sequencing.
Broader Context: Platform Strengths and Limitations in Chemogenomics
Beyond the direct MIP comparison, understanding the inherent strengths and weaknesses of each sequencing technology is essential for application in chemogenomic research.
Table 2: Inherent Characteristics of Illumina and Oxford Nanopore Sequencing Technologies
| Characteristic | Illumina | Oxford Nanopore Technologies (ONT) |
|---|---|---|
| Primary Technology | Short-read, sequencing-by-synthesis | Long-read, electronic nanopore sensing |
| Typical Read Length | 100-300 bp [10] | Thousands to millions of bases (long reads) [10] |
| Key Strengths | Very high base-level accuracy, high throughput, mature bioinformatics tools | Rapid turnaround time (real-time data), portability, ability to detect base modifications |
| Key Limitations | Short reads struggle with complex repeats and structural variants; longer library prep times for some workflows | Higher raw error rate that can affect variant calling [10]; requires specific noise models for low-frequency variant detection [108] |
| Ideal for MIP Panels | Applications requiring the highest possible SNP detection accuracy and low-frequency variant calling [108] | Applications where speed, portability, or long-read phasing across distant targets is beneficial |
The higher error rate of ONT, as noted in Table 2, can have practical consequences. For instance, in a whole-genome study of C. difficile, Nanopore sequences exhibited an average of 640 base errors per genome, which led to the incorrect assignment of over 180 alleles in a core genome MLST analysis, rendering the phylogeny less accurate than the Illumina equivalent for investigating transmission events [10]. However, for MIP-based targeted detection, where amplicons are small and defined, these errors can be mitigated through sufficient coverage and robust bioinformatic pipelines, as evidenced by the strong performance shown in Table 1 [109].
Essential Research Reagent Solutions
The successful implementation of MIP-based sequencing relies on a suite of specialized reagents and tools. The following table details key solutions used in the featured experiments and the broader field.
Table 3: Key Research Reagent Solutions for MIP-Based Targeted Sequencing
| Reagent / Solution | Function in Workflow | Examples & Notes |
|---|---|---|
| Custom MIP Panels | Target enrichment by specifically hybridizing to and capturing genomic regions of interest. | Designed via tools like DesignStudio [111] or MIPgen [108]. Illumina Custom Enrichment Panel v2 uses 120 bp double-stranded probes [111]. |
| High-Fidelity DNA Polymerase | Catalyzes the gap-filling step during circularization with high accuracy to minimize PCR-introduced errors. | Q5 High-Fidelity (used in iMIP) [108] and Phusion HF (used in standard MIP) are common choices. |
| Thermostable Ligase | Seals the nicks in the circularized MIP probe after gap filling. | Ampligase is specifically designed for this purpose and is used in both standard and iMIP protocols [108]. |
| Exonucleases | Digests linear, un-circularized DNA molecules post-ligation, enriching the library for successfully captured targets. | A combination of Exonuclease I and III is standard [108]. The iMIP protocol doubled the enzyme concentration for a faster clean-up. |
| Bioinformatic Tools for Low-VAF | Statistical and machine learning models to distinguish true low-frequency variants from sequencing artifacts. | Essential for mosaic mutation detection [110]. The MIPP-Seq pipeline uses independent amplicon analyses to validate allelic fractions as low as 0.025% [110]. |
This direct comparison demonstrates that MIP panels are a versatile and robust technology for pathogen detection on both Illumina and Oxford Nanopore sequencing platforms. The choice between them is not a matter of which is universally superior, but which is optimal for a specific research context.
- For applications demanding the highest possible accuracy for single nucleotide variant calling, low-frequency mutation detection, and high-resolution phylogenetic analysis, Illumina remains the preferred choice, as evidenced by its higher concordance in pathogen identification [109].
- For applications where speed, portability, and lower initial cost are the primary drivers, and where slightly lower base-level accuracy is an acceptable trade-off, Oxford Nanopore presents a compelling alternative. Its performance with MIP panels is sufficient for reliable pathogen detection and classification in many clinical and public health scenarios [109] [10].
The ongoing development of optimized protocols, such as iMIP, and sophisticated bioinformatic models for noise reduction continues to enhance the performance of MIPs on both platforms, solidifying their role in modern pathogen surveillance and chemogenomic research.
Conclusion
The choice between Illumina and Nanopore sequencing is not a matter of superiority but of strategic alignment with specific chemogenomic objectives. Illumina remains the gold standard for high-throughput, high-accuracy applications requiring broad microbial surveys and exceptional reproducibility. In contrast, Nanopore technology offers unparalleled advantages for real-time analysis, resolving complex genomic structures, and achieving species-level taxonomic resolution, which is critical for understanding antimicrobial resistance and precise microbiome shifts. Future directions point toward the increased use of hybrid sequencing approaches and continuous improvements in bioinformatics and chemistry, particularly for Nanopore, which is rapidly closing the accuracy gap. For the field of chemogenomics, this evolution promises more comprehensive genomic insights, accelerating the discovery of novel therapeutic targets and personalized treatment strategies. Researchers are advised to base their platform selection on a clear understanding of their specific questions, weighing the need for speed, resolution, depth, and accuracy against project constraints and goals.
References
- 1. Sequencing Technology | Sequencing by synthesis [https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology.html]
- 2. Benefits of SBS Technology ǀ Robust sequencing data ... [https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology/sbs-benefits.html]
- 3. Nanopore sequencing accuracy [https://nanoporetech.com/platform/accuracy]
- 4. Nanopore vs Illumina Sequencing Technologies [https://www.cosmosid.com/blog/battle-of-the-sequencers-nanopore-vs-illumina-sequencing-technologies/]
- 5. Comparative analysis of illumina and oxford nanopore ... [https://www.nature.com/articles/s41598-025-18768-3]
- 6. iSeq 100 Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/iseq/specifications.html]
- 7. Comparison of Illumina and Oxford Nanopore Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12179503/]
- 8. NovaSeq 6000 Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html]
- 9. High-performance protocol for ultra-short DNA sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12040124/]
- 10. Comparison of Illumina and Oxford Nanopore sequencing ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11267-9]
- 11. How nanopore sequencing works [https://nanoporetech.com/platform/technology]
- 12. How Nanopore - Long Works — In One Simple... Reads Sequencing [https://www.linkedin.com/pulse/how-nanopore-long-reads-sequencing-works-one-simple-1ip9f]
- 13. London Calling 2025 Technology Update [https://nanoporetech.com/news/london-calling-2025-technology-update]
- 14. A systematic benchmark of Nanopore long-read RNA ... [https://www.nature.com/articles/s41592-025-02623-4]
- 15. disease with only a single molecule | ScienceDaily Detecting [https://www.sciencedaily.com/releases/2025/01/250102162248.htm]
- 16. Targeted nanopore by real-time... | Nature Biotechnology sequencing [https://www.nature.com/articles/s41587-020-0731-9?error=cookies_not_supported&code=bbde0ecf-a316-4ec3-a0e0-82cdb671911e]
- 17. Oxford Nanopore at ASHG 2025 [https://nanoporetech.com/about/events/conferences/ashg-2025]
- 18. Comparison of the performance of MiSeq and NovaSeq in oral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11028001/]
- 19. Illumina advances NovaSeq X Series, delivering single- ... [https://www.illumina.com/company/news-center/press-releases/press-release-details.html?newsid=bdd28781-e0bd-40b1-ad75-9653edee907f]
- 20. Data analysis [https://nanoporetech.com/document/data-analysis]
- 21. A new compression strategy to reduce the size of nanopore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12212073/]
- 22. Comparison of Illumina and Oxford Nanopore sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11783910/]
- 23. MiSeq Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html]
- 24. PacBio vs Oxford Nanopore: Which Long-Read ... [https://www.cd-genomics.com/resource-pacbio-oxford-nanopore-comparison.html]
- 25. MiSeq i100 Series Specifications [https://www.illumina.com/systems/sequencing-platforms/miseq-i100/specifications.html]
- 26. Price List - Nanopore store [https://store.nanoporetech.com/priceList.html]
- 27. Comparison of Illumina and Oxford Nanopore Technology ... [https://pubmed.ncbi.nlm.nih.gov/40434130/]
- 28. Press Release [https://investor.illumina.com/news/press-release-details/2025/Illumina-whole-genome-sequencing-provides-greater-insight-into-genetic-signals-behind-common-diseases--according-to-Nature-study/default.aspx]
- 29. Gene Target Identification in Complex Disease Research [https://www.illumina.com/areas-of-interest/complex-disease-genomics/gene-target-identification.html]
- 30. Science unlocked: publication picks from October 2025 [https://nanoporetech.com/blog/science-unlocked-publication-picks-from-october-2025]
- 31. Evaluation of Illumina and Oxford Nanopore Sequencing ... [https://www.mdpi.com/1422-0067/26/9/4198]
- 32. Illumina fuels multiomic discovery with launch of 5-base ... [https://www.illumina.com/company/news-center/press-releases/2025/ed906867-8ddf-4219-b15f-b8aa8facbd11.html]
- 33. Review Applications of Oxford Nanopore Technology in the ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389425027438]
- 34. The applications and advantages of nanopore sequencing ... [https://www.nature.com/articles/s44259-025-00157-5]
- 35. Communities of plasmids as strategies for antimicrobial ... [https://www.nature.com/articles/s44259-025-00151-x]
- 36. Comparative analysis of illumina and oxford nanopore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12480654/]
- 37. Overcoming challenges in metagenomic AMR surveillance ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1614301/full]
- 38. Nanopore Sequencing-Driven Mapping of Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382671/]
- 39. Comparative analysis of shotgun metagenomics and 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10629391/]
- 40. Comparative Analysis of 16S rRNA Gene and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.670336/full]
- 41. Mock community taxonomic classification performance of ... [https://www.nature.com/articles/s41597-023-02877-7]
- 42. Benchmarking second and third-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9652401/]
- 43. 16S rRNA and metagenomic shotgun sequencing data ... [https://www.nature.com/articles/s41598-022-07995-7]
- 44. Workflow overview: 16S sequencing [https://nanoporetech.com/resource-centre/workflow-16s-sequencing]
- 45. Standardization of 16S rRNA gene sequencing using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11922894/]
- 46. Prospective Comparison Between Shotgun Metagenomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9020828/]
- 47. Oxford Nanopore at ASM Microbe 2025 [https://nanoporetech.com/about/events/conferences/asm-microbe-2025]
- 48. Microbiome sequencing [https://nanoporetech.com/applications/research-areas/microbiome]
- 49. A comparison between full-length 16 rRNA Oxford S ... nanopore [https://pubmed.ncbi.nlm.nih.gov/38713383/]
- 50. Nanopore full length 16S rRNA gene sequencing ... [https://www.nature.com/articles/s41598-025-10999-8]
- 51. A comparison between full-length 16 rRNA Oxford S ... nanopore [https://link.springer.com/article/10.1007/s00203-024-03985-7]
- 52. Evaluation of 16S rRNA gene sequencing for species and ... [https://www.nature.com/articles/s41467-019-13036-1]
- 53. A comparison between full-length 16S rRNA Oxford nanopore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11076400/]
- 54. Evaluation of V3–V4 and FL-16S rRNA amplicon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12379586/]
- 55. The clinical utility of Nanopore 16S rRNA gene sequencing ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1324494/full]
- 56. Comparative evaluation of sequencing platforms: Pacific ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1633360/full]
- 57. Advantages of real-time sequencing [https://nanoporetech.com/platform/technology/advantages-of-real-time-sequencing]
- 58. The performance of nanopore sequencing in rapid ... [https://www.sciencedirect.com/science/article/abs/pii/S0732889325000434]
- 59. Comparative analysis of Illumina, PacBio, and nanopore ... [https://www.frontiersin.org/journals/microbiomes/articles/10.3389/frmbi.2025.1587712/full]
- 60. Science unlocked: publication picks from September 2025 [https://nanoporetech.com/blog/science-unlocked-publication-picks-from-september-2025]
- 61. Nanopore vs. Illumina: Which DNA sequencing technology ... [https://mynucleus.com/blog/nanopore-vs-illumina]
- 62. Hybrid Sequencing: The Future of Accurate and Complete ... [https://strandls.com/articles/hybrid-sequencing-the-future-of-accurate-and-complete-genome-assembly/]
- 63. How does the efficiency of Nanopore compare to Illumina ... [https://www.sciencedirect.com/science/article/pii/S2772735125000666]
- 64. A simplified hybrid capture approach retains high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403294/]
- 65. viralrecon: Introduction [https://nf-co.re/viralrecon/2.6.0/]
- 66. Arcadia-Science/metagenomics: A Nextflow workflow ... [https://github.com/Arcadia-Science/metagenomics]
- 67. DADA2 Pipeline Tutorial (1.16) [https://benjjneb.github.io/dada2/tutorial.html]
- 68. DADA2: High resolution sample inference from Illumina ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4927377/]
- 69. Primed and ready: nanopore metabarcoding can now recover ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10767-4]
- 70. Sequencing DNA with nanopores: Troubles and biases [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0257521]
- 71. Identifying and correcting repeat-calling errors in nanopore ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02751-6]
- 72. Performance of Nanopore and Illumina Metagenomic ... [https://pubmed.ncbi.nlm.nih.gov/36299531/]
- 73. PRONAME: a user-friendly pipeline to process long-read ... [https://pubmed.ncbi.nlm.nih.gov/39758955/]
- 74. Quality Scores for Next Generation Sequencing [https://knowledge.illumina.com/instrumentation/general/instrumentation-general-reference_material-list/000006557]
- 75. Oxford Nanopore announces breakthrough technology ... [https://nanoporetech.com/news/oxford-nanopore-announces-breakthrough-technology-performance-to-deliver-complete-human-genomes-and-richer-multiomic-data-in-london-calling-tech-update]
- 76. Illumina complete long read assay yields contiguous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363240/]
- 77. Comparison of Illumina and Oxford Nanopore sequencing ... [https://pubmed.ncbi.nlm.nih.gov/39885402/]
- 78. Accurately assembling nanopore sequencing data of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392509/]
- 79. Comparative Evaluation of Open-Source Bioinformatics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11680378/]
- 80. Comparison of commonly used software pipelines for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-11001-x]
- 81. GC bias affects genomic and metagenomic reconstructions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7016772/]
- 82. Biases from Oxford Nanopore library preparation kits and their ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11649-z]
- 83. Genomic GC-Content Affects the Accuracy of 16S rRNA ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2017.01934/full]
- 84. Biases in Illumina transcriptome sequencing caused by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2896536/]
- 85. Sequencing DNA with nanopores: Troubles and biases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8486125/]
- 86. Proof of concept for multiplex amplicon sequencing ... [https://www.nature.com/articles/s41598-022-12613-7]
- 87. NGS Library Preparation [https://www.cd-genomics.com/resource-library-preparation-for-ngs.html]
- 88. Chemistry Technical Document [https://nanoporetech.com/document/chemistry-technical-document]
- 89. Illumina Adapter Sequences [https://support.illumina.com/downloads/illumina-adapter-sequences-document-1000000002694.html]
- 90. Comparison of Oxford Nanopore Technologies and ... [https://www.nature.com/articles/s41598-023-36101-8]
- 91. Preparation of DNA Sequencing Libraries for Illumina ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/dna-sequencing-preparation-illumina.html]
- 92. Library adaptors with integrated reference controls improve ... [https://www.nature.com/articles/s41467-022-34028-8]
- 93. Best practices for library quantification [https://knowledge.illumina.com/library-preparation/general/library-preparation-general-reference_material-list/000003750]
- 94. Benchmarking bacterial taxonomic classification using ... [https://www.nature.com/articles/s41597-024-03672-8]
- 95. Benchmarking taxonomic classifiers with Illumina and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9676057/]
- 96. Evaluation of taxonomic classification and profiling methods ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05103-0]
- 97. Assessing Read Quality, Trimming and Filtering - GitHub Pages [https://cloud-span.github.io/nerc-metagenomics03-qc-assembly/01-QC-quality-raw-reads/index.html]
- 98. Strategies and tools in illumina and nanopore‐integrated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10989838/]
- 99. Full-length 16S rRNA gene sequencing by PacBio improves... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10213-5]
- 100. Taxonomy based performance metrics for evaluating ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2896-0]
- 101. HFTC: a hierarchical fungal taxonomic classification model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12531816/]
- 102. Species abundance information improves sequence ... [https://www.nature.com/articles/s41467-019-12669-6]
- 103. Frontiers | High- Resolution Metagenomics of Human Gut Microbiota... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.801587/full]
- 104. Nanopore versus Illumina to study the gut bacterial diversity of ... [https://bmcvetres.biomedcentral.com/articles/10.1186/s12917-025-04693-0]
- 105. Alpha & beta diversity [https://help.partek.illumina.com/partek-flow/user-manual/task-menu/metagenomics/alpha-and-beta-diversity]
- 106. Microbiome differential abundance methods produce ... [https://www.nature.com/articles/s41467-022-28034-z]
- 107. Genomic GC bias correction improves species abundance ... [https://www.nature.com/articles/s41467-025-65530-4]
- 108. An improved molecular inversion probe based targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8826764/]
- 109. Comparison of Illumina and Oxford Nanopore Sequencing ... [https://www.sciencedirect.com/science/article/pii/S1525157822000101]
- 110. MIPP-Seq: ultra-sensitive rapid detection and validation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7881461/]
- 111. Illumina Custom Enrichment Panel v2 [https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/custom-enrichment-panels.html]