CGF vs. MLST: A Comparative Analysis of Bacterial Subtyping Performance for Outbreak Detection and Surveillance
This article provides a comprehensive comparison of Comparative Genomic Fingerprinting (CGF) and Multi-Locus Sequence Typing (MLST) for bacterial subtyping, tailored for researchers and public health professionals.
CGF vs. MLST: A Comparative Analysis of Bacterial Subtyping Performance for Outbreak Detection and Surveillance
Abstract
This article provides a comprehensive comparison of Comparative Genomic Fingerprinting (CGF) and Multi-Locus Sequence Typing (MLST) for bacterial subtyping, tailored for researchers and public health professionals. We explore the foundational principles of each method, detailing their transition from traditional to whole-genome sequencing-based applications. The methodological comparison covers discriminatory power, epidemiological concordance, and practical implementation through available software tools. We address common troubleshooting scenarios and optimization strategies for handling mixed samples and variable sequencing coverage. Finally, we synthesize validation studies and performance metrics from real-world outbreak investigations to guide method selection for specific research and surveillance objectives, underscoring the pivotal role of advanced subtyping in modern epidemiology.
Understanding the Core Principles: From Housekeeping Genes to Genomic Fingerprints
Multi-locus sequence typing (MLST) has stood as a cornerstone technique in molecular epidemiology since its introduction in 1998, providing a standardized, portable approach for the precise identification and differentiation of bacterial strains [1] [2]. This method revolutionized bacterial typing by moving from fragment-based sizing to unambiguous DNA sequence analysis, enabling robust interlaboratory comparisons and global surveillance of bacterial pathogens. By focusing on the nucleotide sequences of approximately seven carefully selected housekeeping genes—essential genes required for basic cellular functions—MLST generates unique allele profiles that facilitate accurate strain identification and in-depth evolutionary analysis [3]. The stability and conservation of these genetic loci provide the foundation for a typing system that balances sufficient variation for discrimination with enough conservation to reveal meaningful evolutionary relationships, establishing MLST as the historical "gold standard" against which newer methods are often measured, particularly in the context of epidemiological research and outbreak investigations.
The MLST Methodology: A Detailed Workflow
The conventional MLST process follows a meticulously defined pathway to transform bacterial isolates into comparable sequence types (STs). The workflow can be broken down into several critical stages, each contributing to the method's renowned reproducibility.
Experimental Protocol
The wet-lab procedure begins with the preparation of high-quality genomic DNA from bacterial isolates, requiring a total amount >500 ng and a concentration >10 ng/μL, with optimal purity (OD260/280 ratio between 1.8 and 2.0) and no degradation or contamination [3]. The subsequent steps are:
- PCR Amplification: Using sequence-specific primers, each of the seven housekeeping gene loci is amplified via polymerase chain reaction (PCR). The primer sets are designed to target internal fragments of approximately 400-600 base pairs in length [3] [1].
- Purification and Sequencing: The PCR amplicons are purified to remove enzymes, primers, and nucleotides that could interfere with sequencing. Historically, this has been performed using Sanger sequencing technology with BigDye Terminator chemistry on platforms such as the ABI 3100 or 3730 DNA analyzers [4].
- Sequence Assembly and Quality Control: The resulting sequences for each locus are assembled and checked for errors and overall quality using specialized software such as the SeqMan program within the Lasergene suite [4].
Data Analysis and Sequence Type Assignment
The bioinformatics phase translates raw sequence data into a standardized genotype:
- Allele Assignment: For each of the seven loci, the determined sequence is compared against a curated database of known alleles (e.g., on PubMLST). If the sequence perfectly matches a known allele, it is assigned the corresponding allele number. If it is a novel sequence, a new allele number is issued by the database [2].
- Profile and ST Determination: The combination of the seven allele numbers forms the isolate's allelic profile. This unique profile is then mapped to a Sequence Type (ST). Each unique profile receives a unique ST number, creating a portable and unambiguous identifier for the strain [3] [2].
- Advanced Analysis: Further bioinformatics analyses can include identifying polymorphic sites, population structure analysis, phylogenetic analysis, and recombination analysis, which help elucidate the genetic relationships and evolutionary history of the strains under investigation [3].
The following diagram illustrates the complete workflow from isolate to final sequence type.
MLST in a Evolving Typing Landscape: Comparison with CGF
While MLST has been a foundational tool, the field of molecular epidemiology continues to advance, leading to the development of new methods with higher resolution. One such method is Comparative Genomic Fingerprinting (CGF), which provides a contrasting approach to bacterial subtyping. The table below summarizes the core differences between these two techniques.
Table 1: Fundamental comparison between MLST and CGF
| Feature | Multi-Locus Sequence Typing (MLST) | Comparative Genomic Fingerprinting (CGF) |
|---|---|---|
| Genetic Target | Nucleotide sequence of ~7 core housekeeping genes [3] | Presence/absence of ~40 accessory genomic genes [4] [5] |
| Basis of Discrimination | Sequence variation (point mutations) in conserved genes [3] | Variation in gene content (insertions/deletions) in the accessory genome [4] |
| Primary Application | Long-term epidemiological and population studies, evolutionary analysis [4] [3] | Short-term outbreak investigations and high-resolution surveillance [4] [5] |
| Key Advantage | High portability, excellent for interlab comparisons and building global databases [4] [6] | Higher discriminatory power for distinguishing closely related isolates [4] |
| Typical Technology | Sanger sequencing [3] | Multiplex PCR [4] |
Quantitative Performance Comparison
The theoretical distinctions between MLST and CGF translate into measurable differences in performance. A validation study on Campylobacter jejuni directly compared the two methods using a set of 412 isolates from various sources. The key quantitative findings are summarized in the table below.
Table 2: Performance comparison of CGF40 and MLST for C. jejuni subtyping [4]
| Method | Simpson's Index of Diversity (ID) | Clonal Complex (CC) ID | Sequence Type (ST) ID | Concordance with Reference Phylogeny |
|---|---|---|---|---|
| CGF40 | 0.994 | - | - | High (CGF and MLST are highly concordant) [4] |
| MLST | - | 0.873 | 0.935 | High [6] |
The significantly higher Simpson's index of diversity for CGF40 highlights its superior discriminatory power compared to MLST, which sometimes lacks the resolution needed for short-term investigations [4]. This enhanced resolution allows CGF to differentiate between closely related isolates that share an identical MLST Sequence Type, a capability crucial for pinpointing transmission routes in acute outbreak scenarios [4] [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of MLST and CGF relies on a suite of specific reagents and materials. The following table details the key components required for the core experimental workflows.
Table 3: Essential research reagents and materials for MLST and CGF
| Item | Function/Description | Typical Example/Kit |
|---|---|---|
| DNA Purification Kit | Extracts high-quality genomic DNA from bacterial cultures, free of contaminants that inhibit PCR or sequencing. | QIAamp DNA Mini Kit [7], PureGene Genomic DNA Purification Kit [4] |
| PCR Primers | Sequence-specific oligonucleotides designed to amplify the target loci (7 housekeeping genes for MLST; ~40 accessory genes for CGF). | Custom-designed primers [4] [7] |
| PCR Master Mix | A pre-mixed solution containing DNA polymerase, dNTPs, MgCl₂, and buffers for efficient amplification of target genes. | Not specified in results, but essential for workflow. |
| PCR Purification Kit | Removes excess primers, enzymes, and dNTPs from PCR products prior to sequencing. | Montage PCR Centrifugal Filter Devices [4], QIAquick PCR Purification Kit [7] |
| Sequencing Chemistry | Fluorescently labeled di-deoxy terminators used in cycle sequencing reactions. | BigDye Terminator 3.1 [4] |
| Genetic Analyzer | Capillary electrophoresis instrument for separating and detecting fluorescently labeled DNA fragments. | ABI 3100 or 3730 DNA Analyzer [4] |
| Bioinformatics Software | For sequence assembly, quality control, allele calling, and phylogenetic analysis. | SeqMan (Lasergene) [4], BLAST [3] |
Advancements and Future Directions: The Evolution Beyond 7 Genes
The principle of multi-locus analysis established by MLST has evolved with technological progress. The advent of next-generation sequencing (NGS) has facilitated the development of more powerful, genome-scale typing methods [3] [8].
- Core Genome MLST (cgMLST): This approach expands the MLST concept to include hundreds of genes present in the core genome of a species, providing significantly higher resolution and discriminatory power than traditional 7-gene MLST [8] [1]. It is particularly valuable for distinguishing highly clonal outbreak strains.
- Whole Genome MLST (wgMLST): wgMLST goes a step further by analyzing variation across both the core and accessory genome, offering the highest possible resolution for strain typing by effectively comparing nearly the entire gene repertoire [3] [1].
- Whole Genome Sequencing (WGS) as the Ultimate Gold Standard: WGS is increasingly considered the new benchmark for microbial characterization. It provides a complete dataset from which any molecular type (MLST, cgMLST, CGF) can be derived in silico, and it enables the most comprehensive phylogenetic analyses and transmission tracking [6] [8].
The following diagram illustrates the logical and technological progression from traditional MLST to these more advanced genome-based typing methods.
MLST, with its foundation in the sequences of seven housekeeping genes, has earned its status as a gold standard in bacterial typing through its standardization, portability, and profound contribution to our understanding of bacterial population genetics and epidemiology. However, the escalating demands of public health surveillance and outbreak investigation for ever-greater resolution are steadily shifting the field toward genome-based methods like cgMLST and WGS. In this evolving landscape, methods like CGF have demonstrated that targeting the accessory genome can provide a high-resolution, highly deployable alternative for specific short-term applications. Therefore, while the seven genes of MLST remain a fundamental and historically crucial tool, the future of bacterial subtyping lies in the comprehensive and unparalleled power of the entire genome.
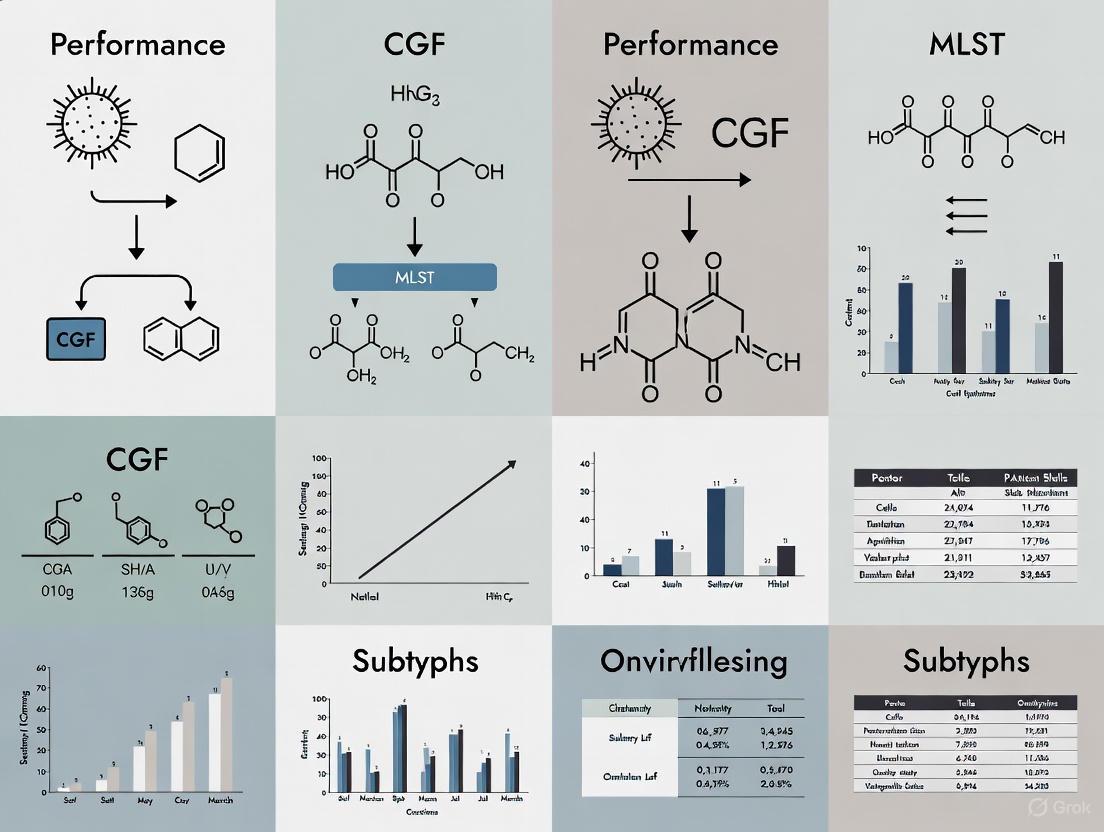
In the ongoing battle against bacterial pathogens, accurate strain typing is crucial for effective surveillance and outbreak investigations. Molecular subtyping methods allow researchers to differentiate bacterial isolates beyond the species level, enabling the tracking of contamination sources and the identification of transmission pathways. For decades, methods like Multilocus Sequence Typing (MLST) have served as valuable tools, but they can lack the resolution needed for short-term epidemiological investigations. To address this gap, Comparative Genomic Fingerprinting (CGF) emerges as a rapid, high-resolution multiplex PCR approach that targets variable genomic regions, offering enhanced discriminatory power for bacterial subtyping.
What is Comparative Genomic Fingerprinting (CGF)?
Comparative Genomic Fingerprinting is a multiplex PCR-based method that exploits genetic variability in the accessory genome content of bacteria. Unlike MLST, which sequences segments of a few (typically seven) housekeeping genes, CGF simultaneously targets multiple loci (e.g., 40 genes in the CGF40 assay) distributed across the genome that demonstrate presence/absence variation among strains. This approach captures strain-to-strain relationships inferred from whole-genome comparative analysis, providing a higher-resolution fingerprint at a lower cost and faster turnaround than whole-genome sequencing.
The development of a CGF assay involves careful selection of target genes based on specific criteria: they should be accessory genes (absent in some strains), represent unbiased genes with adequate carriage across populations, be distributed across hypervariable genomic regions, and enable the reproduction of strain relationships seen in whole-genome analyses [4].
Head-to-Head: CGF Versus MLST Performance
Extensive validation studies have directly compared CGF with MLST, revealing important performance differences that researchers must consider when selecting a subtyping method.
Table 1: Comparative Performance of CGF and MLST for Bacterial Subtyping
| Parameter | CGF (CGF40 Assay) | MLST | Implications for Research |
|---|---|---|---|
| Discriminatory Power | Higher (Simpson's ID = 0.994) [4] | Lower (Simpson's ID = 0.935 for ST) [4] | CGF better differentiates closely related isolates within the same ST |
| Methodology Basis | Presence/absence of 40 accessory genes via multiplex PCR [4] | Nucleotide sequences of ~7 housekeeping genes [4] | CGF captures more genomic diversity; MLST focuses on core genome |
| Concordance with MLST | High (Wallace coefficient supports high concordance) [4] | N/A | CGF maintains phylogenetic relationships identified by MLST |
| Cost & Speed | Rapid, lower cost [4] | More expensive, slower [9] | CGF more suitable for high-throughput or resource-limited settings |
| Epidemiological Resolution | Superior for short-term investigations [4] | Better for long-term evolutionary studies [4] | CGF ideal for outbreak tracking; MLST for population genetics |
The significantly higher Simpson's index of diversity values obtained with CGF40 highlights its enhanced ability to distinguish between closely related bacterial isolates. This is particularly valuable for differentiating highly prevalent sequence types such as ST21 and ST45 in Campylobacter jejuni, where MLST may lack sufficient resolution [4]. Despite this higher discrimination, CGF and MLST show high concordance, meaning that CGF maintains the broader phylogenetic relationships established by MLST while providing additional resolution.
Inside the Black Box: Experimental Protocols and Methodologies
CGF Assay Workflow
The technical implementation of CGF involves a structured process from gene selection to data analysis, each step critical to ensuring reproducible, high-quality results.
Table 2: Key Research Reagent Solutions for CGF Implementation
| Reagent/Equipment | Function in CGF Protocol | Implementation Example |
|---|---|---|
| Multiplex PCR Primers | Simultaneous amplification of multiple target loci | 40 gene-specific primers pooled in 8 multiplex reactions [4] |
| DNA Purification Kit | High-quality genomic DNA extraction | PureGene genomic DNA purification kit [4] |
| PCR Enzymes/Master Mix | Amplification of target genes | Phusion High-fidelity DNA Polymerase [10] |
| Thermal Cycler | Precise temperature cycling for PCR | Standard PCR thermal cycler |
| Electrophoresis System | Size separation of amplification products | Agarose gel electrophoresis or microfluidic chips |
Diagram 1: CGF experimental workflow for bacterial subtyping.
MLST Methodology
In contrast to CGF, MLST follows a different analytical pathway focused on sequence-based typing of core housekeeping genes:
Diagram 2: MLST methodology based on housekeeping gene sequencing.
The fundamental difference lies in what each method detects: CGF identifies the presence or absence of accessory genes through amplification pattern analysis, while MLST identifies sequence variations in core housekeeping genes through nucleotide sequencing [4].
Contextualizing CGF Within the Broader Molecular Typing Landscape
While CGF shows superior performance compared to MLST, it's important to understand how it fits alongside other typing methods used in modern microbiology laboratories.
Table 3: CGF Positioned Among Current Bacterial Typing Methods
| Method | Resolution | Turnaround Time | Cost | Primary Application |
|---|---|---|---|---|
| CGF | High | Days | $$ | Outbreak investigation, source tracking |
| MLST | Moderate | Days-Weaks | $$ | Population studies, long-term epidemiology |
| PFGE | Moderate | 3-4 days | $$ | Outbreak investigation (historical gold standard) |
| rep-PCR | High | <4 hours [9] | $ | Rapid screening, local surveillance |
| cgMLST | Very High | Weeks | $$$ | High-resolution outbreak investigation |
| WGS | Highest | Weeks | $$$$ | Comprehensive genetic analysis |
This comparison reveals CGF as a balanced option offering high resolution with moderate cost and time requirements, positioned between rapid but lower-resolution methods like rep-PCR and comprehensive but resource-intensive approaches like whole-genome sequencing (WGS) [8] [11].
The choice between CGF and MLST ultimately depends on the specific research question and practical constraints. CGF offers clear advantages when high discriminatory power is needed for short-term epidemiological investigations, such as outbreak detection and source tracking, particularly for highly diverse pathogens like Campylobacter jejuni [4]. Its cost-effectiveness and rapid turnaround make it deployable for routine surveillance.
MLST remains valuable for long-term epidemiological studies, evolutionary analysis, and global comparisons, as its sequence-based data are highly portable and standardized [4] [11]. The established MLST databases facilitate international collaboration and clone recognition.
For comprehensive surveillance programs, a tiered approach may be most effective: using CGF for high-resolution screening of potential outbreaks and MLST for placing isolates into global context. As sequencing costs continue to decline, WGS-based methods are becoming more accessible, but CGF remains a powerful, cost-effective tool for laboratories requiring high-resolution subtyping without the bioinformatics burden of whole-genome analysis.
The field of bacterial molecular subtyping has undergone a revolutionary shift with the advent of whole-genome sequencing (WGS). This transition has enabled the development of highly discriminatory in silico typing methods that are transforming outbreak investigation, pathogen surveillance, and phylogenetic studies. Among these methods, in silico Multilocus Sequence Typing (MLST) and Comparative Genomic Fingerprinting (CGF) represent two powerful approaches that leverage WGS data to provide unprecedented resolution for strain differentiation [4] [11]. This guide objectively compares the performance, applications, and technical requirements of these methods, providing researchers with experimental data and protocols to inform their selection of subtyping approaches for bacterial pathogen research.
Methodological Foundations and Principles
In Silico Multilocus Sequence Typing (MLST)
Traditional MLST schemes characterize bacterial isolates based on the sequences of approximately seven housekeeping genes, assigning unique sequence types (STs) based on allele profiles [11] [12]. The in silico adaptation of this method extracts these allele sequences directly from WGS data, maintaining backward compatibility with established MLST databases while dramatically reducing turnaround time. This approach preserves the standardized nomenclature and global classification system that has made MLST invaluable for long-term epidemiological studies and population genetics [12]. Core genome MLST (cgMLST) expands this concept by utilizing hundreds to thousands of core genes distributed across the genome, offering significantly enhanced discriminatory power while maintaining the benefits of a standardized, portable nomenclature system [8] [13] [12].
Comparative Genomic Fingerprinting (CGF)
Comparative Genomic Fingerprinting represents a different philosophical approach, targeting genetic variability in the accessory genome content rather than conserved housekeeping genes. The CGF method employs multiplex PCR or in silico analysis of multiple loci widely distributed around the genome that demonstrate presence-absence variation among strains [4]. For example, the CGF40 assay for C. jejuni utilizes 40 gene targets selected based on five criteria: confirmed absence in one or more isolates from genomic surveys, unbiased distribution across populations, representative genomic distribution, ability to capture strain relationships from whole-genome analysis, and presence in multiple public genomes to enable SNP-free primer design [4]. This strategic selection of accessory gene targets provides resolution for differentiating closely related strains that may be indistinguishable by conventional MLST.
Comparative Performance Analysis
Discrimination Power and Typing Resolution
Multiple studies have quantitatively compared the discriminatory power of these subtyping methods, with CGF generally demonstrating superior resolution for short-term epidemiological investigations.
Table 1: Comparison of Discriminatory Power for Campylobacter jejuni Subtyping
| Typing Method | Simpson's Index of Diversity | Target Loci | Epidemiological Concordance |
|---|---|---|---|
| CGF40 | 0.994 | 40 accessory genes | High for outbreak detection |
| MLST (Sequence Type) | 0.935 | 7 housekeeping genes | Moderate for outbreak detection |
| MLST (Clonal Complex) | 0.873 | 7 housekeeping genes | Lower for outbreak detection |
As evidenced in a study of 412 C. jejuni isolates from various sources, CGF40 exhibited significantly higher discriminatory power than MLST, capable of differentiating within highly prevalent sequence types such as ST21 and ST45 that are challenging to resolve with conventional MLST [4]. The CGF approach effectively captures strain-to-strain relationships inferred from whole-genome comparative genomic analysis, making it particularly valuable for investigating potential outbreak clusters.
For other pathogens, cgMLST has demonstrated resolution comparable to single nucleotide polymorphism (SNP)-based analyses while offering advantages in standardization. In a study of Salmonella enterica serovar Enteritidis, cgMLST analysis was congruent with SNP-based phylogeny and epidemiological data, successfully contextualizing a multi-country outbreak [12]. Similarly, both cgMLST and coreSNP analyses showed superior discrimination compared to PFGE for Klebsiella pneumoniae surveillance, though cgMLST appeared inferior to coreSNP in phylogenetic reconstruction of the CG258 clonal group [8].
Technical Implementation and Workflow Considerations
The practical implementation of these methods varies significantly in terms of technical requirements, turnaround time, and data portability.
Table 2: Technical Comparison of Subtyping Method Implementation
| Parameter | In Silico MLST/cgMLST | Enhanced CGF |
|---|---|---|
| Primary data source | Whole-genome sequencing | Whole-genome sequencing or multiplex PCR |
| Analysis workflow | Assembly-based or read-mapping | Presence/absence calling of target loci |
| Scheme scalability | Highly scalable (7 to >2,000 loci) | Typically fixed (e.g., 40-50 loci) |
| Interlaboratory reproducibility | High with standardized schemes | High with defined gene targets |
| Database infrastructure | PubMLST, EnteroBase | Custom databases |
| Computational requirements | Moderate to high | Moderate |
In silico MLST and cgMLST typically employ either assembly-based approaches using tools like SPAdes, Shovill, or Unicycler, or read-mapping approaches using tools like Mentalist [13] [14]. The assembly-based approach can be impacted by genome composition characteristics such as repetitive sequences, insertion sequences, and GC content, potentially introducing variability in cgMLST results [13]. In contrast, CGF utilizes a more targeted analysis, assessing the presence or absence of predefined accessory gene targets, which can be implemented through PCR or in silico analysis of sequencing data [4].
Experimental Protocols and Validation
CGF40 Assay Development and Validation Protocol
The development and validation of the CGF40 method for C. jejuni provides a template for implementing enhanced CGF approaches:
Step 1: Marker Selection
- Identify prospective marker genes from comparative genomic surveys based on bimodal log ratio distributions indicating clear presence-absence patterns [4]
- Apply population frequency analysis to select unbiased genes with adequate carriage across datasets
- Ensure representative genomic distribution across hypervariable regions
- Verify ability to capture strain relationships inferred from whole-genome analysis
Step 2: Assay Design
- Extract orthologous sequences from available complete and draft genomes using BLAST
- Generate multiple sequence alignments for each set of orthologues using ClustalX
- Design SNP-free PCR primers using Primer3 for compatibility with both laboratory and in silico implementation [4]
- Assemble genes into multiplex PCRs (e.g., 8 multiplex PCRs targeting 5 loci each)
Step 3: Validation
- Compare performance against established typing methods (MLST, PFGE) using appropriate diversity measures
- Calculate Simpson's index of diversity to quantify discriminatory power
- Determine Wallace coefficients to assess concordance with existing methods
- Validate epidemiological concordance using isolates with known relationships [4]
cgMLST Implementation Protocol
For cgMLST implementation, the following protocol ensures reproducible results:
Step 1: Scheme Selection
- Select appropriate cgMLST scheme from public repositories (PubMLST, cgmlst.org)
- For A. baumannii, schemes containing 2,390 loci have been successfully implemented [14]
- For Salmonella enterica, schemes with 500-3,000 loci provide sufficient resolution
Step 2: Data Processing
- Perform quality control on sequencing reads using FastQC and multiQC [15] [14]
- Assemble genomes using optimized pipelines (SPAdes, Shovill, or Unicycler)
- Assess assembly quality using CheckM2 for completeness and contamination [14]
Step 3: Allele Calling
- Utilize chewBBACA or similar software for allele calling against the selected scheme [13] [14]
- Apply appropriate quality thresholds for allele calling
- Export allelic profiles for cluster analysis
Step 4: Cluster Analysis
- Use ReporTree with MSTreeV2 method for clustering at defined allelic difference thresholds (e.g., ≤9 alleles for closely related isolates) [14]
- Construct minimum spanning trees using GrapeTree
- Validate clusters with epidemiological data
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Computational Tools for Bacterial Subtyping
| Item | Function | Example Products/Tools |
|---|---|---|
| DNA extraction kits | High-quality genomic DNA isolation | QIAamp DNA Mini Kit, Maxwell 16 Cell DNA Purification Kit |
| Whole-genome sequencing platforms | Generate raw sequence data | Illumina NextSeq500, NovaSeq 6000; PacBio |
| Assembly tools | Reconstruct genomes from sequencing reads | SPAdes, Shovill, Unicycler |
| cgMLST analysis software | Allele calling and profile generation | chewBBACA, Ridom SeqSphere+ |
| CGF analysis tools | Presence/absence calling of target loci | Custom scripts, BLAST-based pipelines |
| Typing databases | Scheme storage and profile comparison | PubMLST, EnteroBase, cgmlst.org |
| Phylogenetic visualization | Tree construction and annotation | GrapeTree, iTOL |
Analysis Workflow and Technical Pathways
The following diagram illustrates the comparative workflows for implementing in silico MLST and enhanced CGF methods:
The transition to whole-genome sequencing has fundamentally transformed bacterial subtyping, enabling the development of highly discriminatory in silico methods like MLST/cgMLST and enhanced CGF. The experimental data and performance comparisons presented in this guide demonstrate that CGF generally offers superior discriminatory power for outbreak investigations and short-term epidemiological studies, particularly for genetically diverse pathogens like Campylobacter jejuni [4]. In contrast, in silico MLST and cgMLST provide excellent resolution for population studies and long-term epidemiology while maintaining standardized nomenclature essential for global surveillance [11] [12].
The choice between these methods ultimately depends on research objectives, technical resources, and the specific pathogen under investigation. For outbreak investigations requiring high resolution among closely related strains, CGF approaches provide exceptional discriminatory power. For broader population studies and surveillance programs, cgMLST offers an optimal balance of resolution, standardization, and data portability. As WGS continues to become more accessible and bioinformatics tools further mature, both approaches will play increasingly important roles in public health microbiology and bacterial pathogen research.
Molecular subtyping of bacterial pathogens is a cornerstone of modern public health epidemiology, enabling outbreak detection, source tracking, and evolutionary studies. For years, Multi-Locus Sequence Typing (MLST) has served as the gold standard, providing a portable and reproducible system for classifying bacterial strains based on the sequences of a limited set of housekeeping genes [16]. However, the advent of whole-genome sequencing (WGS) has facilitated the development of high-resolution methods, including Comparative Genomic Fingerprinting (CGF) and core genome MLST (cgMLST), which offer significantly enhanced discrimination between closely related bacterial isolates [4] [17]. This guide provides an objective comparison of the performance of CGF and MLST, focusing on the critical metrics of discriminatory power and epidemiological concordance, to inform researchers and public health professionals in selecting appropriate subtyping tools for outbreak investigations and surveillance.
Performance Comparison: CGF vs. MLST
The efficacy of a subtyping method is primarily evaluated based on its ability to distinguish between unrelated strains (discriminatory power) and its capacity to correctly group isolates from a common outbreak (epidemiological concordance). The table below summarizes a direct, quantitative comparison between a 40-gene CGF assay (CGF40) and standard MLST for Campylobacter jejuni [4].
Table 1: Quantitative Performance Comparison of CGF40 and MLST for Campylobacter jejuni Subtyping
| Performance Metric | CGF40 | MLST (Sequence Type) | MLST (Clonal Complex) |
|---|---|---|---|
| Simpson's Index of Diversity | 0.994 | 0.935 | 0.873 |
| Primary Typing Method Wallace Coefficient (Concordance with MLST) | - | 0.82 (to Clonal Complex) | - |
| Secondary Typing Method Wallace Coefficient (Concordance with CGF40) | 0.99 (to Sequence Type) | - | - |
| Principle of Method | Presence/absence of 40 accessory genes | Nucleotide sequences of 7 housekeeping genes | Groups of related Sequence Types |
The data demonstrates that CGF40 exhibits a higher discriminatory power than MLST, as indicated by its superior Simpson's Index of Diversity (0.994 for CGF40 vs. 0.935 for MLST Sequence Types) [4]. This means CGF40 is more likely to distinguish between two unrelated C. jejuni isolates picked at random from the population. Furthermore, the high Wallace coefficient (0.99) indicates that isolates with an identical CGF40 profile almost always belong to the same MLST sequence type, confirming high concordance between the methods while the CGF40 provides finer resolution [4].
In the context of public health surveillance for outbreak detection, cgMLST (a method conceptually similar to CGF) has been validated for national surveillance systems. For Shiga-toxin producing E. coli (STEC), the U.S. PulseNet system defines a national cluster as five or more clinical cases within 60 days that are related within 0-10 allelic differences based on cgMLST [17]. This high-resolution clustering is crucial for identifying potential outbreaks rapidly and accurately.
Experimental Protocols for Method Evaluation
Protocol for CGF40 Assay and Validation
The development and validation of a CGF assay, as exemplified for C. jejuni, involve a structured bioinformatics and laboratory workflow [4]:
- Marker Selection: Prospective gene targets are identified from comparative genomic surveys based on five criteria:
- Variable presence/absence across isolates (accessory genome).
- Unbiased carriage frequency (avoiding genes that are almost always present or absent).
- Representative genomic distribution across known hypervariable regions.
- Ability to recapitulate strain relationships inferred from whole-genome analysis.
- Conservation in multiple reference genomes to facilitate SNP-free primer design.
- Assay Design: For the CGF40 assay, 40 genes meeting the above criteria were selected. Orthologous sequences from available genomes are aligned, and PCR primers are designed in conserved regions to avoid single-nucleotide polymorphisms (SNPs). The 40 targets are divided into 8 multiplex PCRs, each targeting 5 loci [4].
- Wet-Lab Analysis: Genomic DNA is extracted from bacterial isolates. The multiplex PCRs are run, and the presence or absence of each of the 40 amplicons is scored to generate a unique fingerprint for each isolate.
- Validation and Comparison: The performance of the CGF assay is validated against a established method like MLST. A collection of hundreds of isolates from diverse sources (e.g., agricultural, environmental, retail, clinical) is typed by both methods. Simpson's Index of Diversity is calculated for each method, and Wallace coefficients are determined to measure concordance [4].
Protocol for MLST and cgMLST Analysis
The standard and core-genome MLST workflows are as follows:
- Classical MLST:
- Locus Amplification and Sequencing: Seven designated housekeeping genes are PCR-amplified and sequenced using standard protocols [4] [18].
- Sequence Type Assignment: The sequence for each locus is compared to a curated database (e.g., PubMLST). Each unique sequence is assigned an allele number, and the combination of alleles across the seven loci defines a Sequence Type (ST) [16] [18].
- Clonal Complex Definition: Related STs are grouped into Clonal Complexes (CCs) using algorithms such as eBURST, which identifies groups of STs that share a recent common ancestor [19] [18].
- cgMLST for Outbreak Surveillance (as used in PulseNet 2.0):
- Whole-Genome Sequencing: Isolate genomes are sequenced to high quality.
- Bioinformatics Analysis: The WGS data is processed through a standardized pipeline (e.g., the PulseNet 2.0 workflow) that includes quality assessment, de novo assembly, and allele calling for a scheme of hundreds to thousands of core genes [17].
- Cluster Detection: Genetic relatedness is assessed based on the number of allelic differences across the core genome. For STEC, an outbreak threshold of 0-10 allele differences is used to define a cluster [17].
- Concordance Validation: The performance of cgMLST is validated against other WGS-based methods like high-quality SNP (hqSNP) analysis and whole-genome MLST (wgMLST) by analyzing known outbreak isolates. Parameters such as pairwise genomic differences and clustering concordance (using tanglegrams and indices like Baker's Gamma Index) are evaluated to ensure epidemiological accuracy [17].
Workflow Diagram: Subtyping Method Evaluation
The following diagram illustrates the logical workflow for evaluating and comparing bacterial subtyping methods, from isolate collection to performance metric calculation.
Subtyping Method Evaluation Workflow - This diagram shows the parallel processing of bacterial isolates through MLST and CGF/cgMLST protocols, followed by integrated analysis using epidemiological data to calculate key performance metrics.
Successful implementation and comparison of subtyping methods rely on specific laboratory reagents, bioinformatics tools, and reference databases.
Table 2: Essential Research Reagents and Resources for Bacterial Subtyping Studies
| Category | Item | Function in Subtyping Analysis |
|---|---|---|
| Laboratory Reagents | Commercial DNA extraction kits | High-quality, pure genomic DNA is essential for reliable PCR and sequencing. |
| PCR Master Mix & Primers | For amplification of target loci in MLST and CGF assays. | |
| Sequencing Reagents/Kits | For determining nucleotide sequences of MLST amplicons or whole genomes. | |
| Bioinformatics Tools | BLAST Suite | For comparing sequence data against allele databases for MLST assignment [18]. |
| PYANI | For calculating Average Nucleotide Identity to assess genomic similarity [18]. | |
| GetHomologues/GetPhylomarkers | For identifying core genes and phylogenetic markers from WGS data [18]. | |
| RGI & CARD Database | For in silico prediction of antibiotic resistance genes from WGS data [18]. | |
| Reference Databases | PubMLST | Curated public repository for MLST allele and sequence type definitions [16]. |
| Kaptive | For capsule (K) and lipooligosaccharide (OCL) locus typing from WGS data [18]. | |
| NCBI GenBank/RefSeq | Primary databases for depositing and retrieving whole-genome sequence data. |
The comparative data clearly demonstrates that CGF and related core-genome methods offer superior discriminatory power for bacterial subtyping compared to traditional MLST, while maintaining high epidemiological concordance. This enhanced resolution is critical for detecting and investigating outbreaks, particularly for closely related strains where standard MLST may lack sufficient differentiation. The choice between methods depends on the specific application: MLST remains valuable for long-term phylogenetic and population structure studies, while CGF and cgMLST are better suited for high-resolution outbreak detection and short-term epidemiological investigations. The ongoing integration of these WGS-based methods into national surveillance networks, such as PulseNet 2.0, underscores their reliability and establishes them as the new benchmark for public health pathogen subtyping.
Implementation in the Lab and Field: A Practical Guide to Typing Workflows
The field of bacterial subtyping has been revolutionized by whole-genome sequencing (WGS), enabling a transition from traditional molecular techniques to comprehensive in silico analysis. This shift is particularly relevant in the broader context of comparing core genome MLST (cgMLST) against conventional multi-locus sequence typing (MLST) for bacterial subtyping research. While traditional MLST relies on sequencing 6-8 housekeeping genes, cgMLST expands this to hundreds or thousands of core genes, providing significantly enhanced resolution for outbreak investigation and population studies [20] [21]. Several computational tools have been developed to extract this typing information directly from raw sequencing data, bypassing the need for complete genome assembly. Among these, ARIBA, SRST2, and stringMLST have emerged as prominent solutions, each employing distinct algorithmic approaches with implications for their performance characteristics and suitability for different research scenarios [22] [23]. This review provides a comparative analysis of these three tools, evaluating their methodologies, performance metrics, and practical implementation to guide researchers in selecting the most appropriate solution for their bacterial subtyping needs.
The fundamental algorithmic differences between ARIBA, SRST2, and stringMLST underlie their varied performance in accuracy, speed, and resource consumption.
SRST2: Read Mapping-Based Typing
SRST2 (Short Read Sequence Typing) represents a pioneering read mapping-based approach for gene detection and MLST typing. Its workflow begins by mapping Illumina sequencing reads against reference allele sequences using Bowtie2 with sensitive parameters [24] [25]. Following mapping, SAMtools generates pileups, which SRST2 analyzes using a sophisticated statistical scoring system. This system performs binomial tests at each position in the reference sequence to quantify evidence against the presence of each reference allele, accounting for sequencing error rates. The results are visualized using a quantile-quantile (Q-Q) plot, where the slope of the fitted linear model serves as the allele score [25]. The allele with the lowest score (flattest slope) is identified as the best match, with outliers in the Q-Q plot typically indicating single nucleotide polymorphisms (SNPs) or indels relative to the reference. SRST2 reports the closest matching allele, average read depth, and flags potential novel alleles when exact matches are not found [25] [26].
ARIBA: Local Assembly-Based Typing
ARIBA (Antibiotic Resistance Identification By Assembly) employs a fundamentally different strategy centered on local de novo assembly. Rather than mapping reads directly to reference databases, ARIBA first maps reads to clustered reference sequences using Minimap, then performs local assembly of the mapped reads for each cluster using Fermi-lite [22] [26]. The resulting contigs are aligned to the best-matched reference sequence within each cluster using nucmer from the MUMmer package. This assembly-based approach provides ARIBA with several unique capabilities, including the determination of whether a queried coding sequence is complete and functional, or potentially disrupted by insertions or other structural variations [26]. ARIBA generates comprehensive flags for each allele call, detailing assembly quality and sequence characteristics, and provides functional predictions by distinguishing between synonymous and non-synonymous mutations [26].
stringMLST: k-mer Based Typing
stringMLST represents a third algorithmic paradigm, utilizing exact k-mer matching to completely bypass both read mapping and assembly processes. The tool builds a hash table data structure indexing all k-mers present in the MLST allele database [23] [20]. For each k-mer in the sequencing reads, stringMLST casts "votes" for all alleles containing that k-mer. The allele with the highest vote count for each locus is selected as the best match [20]. This k-mer counting approach eliminates computationally intensive alignment steps, making stringMLST exceptionally fast for traditional MLST schemes. However, this method may not scale efficiently to larger cgMLST schemes containing thousands of genes, a limitation addressed by next-generation k-mer tools like MentaLiST and STing [23] [20].
Table 1: Comparison of Fundamental Algorithmic Approaches
| Tool | Core Algorithm | Key Dependencies | Primary Input | Key Outputs |
|---|---|---|---|---|
| SRST2 | Read mapping + statistical scoring | Bowtie2, SAMtools, SciPy | Raw sequencing reads (paired/single-end) | Best-matching alleles, consensus sequences, coverage metrics |
| ARIBA | Local assembly + contig alignment | Minimap, Fermi-lite, MUMmer, CD-HIT | Paired-end sequencing reads | Best-matching alleles, assembly flags, variant annotations |
| stringMLST | k-mer counting + voting | Custom k-mer index | Raw sequencing reads | Best-matching alleles, allele scores |
Figure 1: Comparative Workflows of MLST Typing Tools
Performance Comparison and Benchmarking
Multiple studies have conducted systematic evaluations of MLST typing tools using both real and simulated datasets, providing insights into the relative performance of ARIBA, SRST2, and stringMLST across various metrics.
Accuracy and Typing Resolution
In comprehensive benchmarking studies, all three tools demonstrate high accuracy when evaluating traditional 7-gene MLST schemes under optimal sequencing conditions. A 2017 comparison of eight MLST software applications against real and simulated data found that SRST2 and ARIBA both achieved high accuracy in calling sequence types from WGS data [22]. SRST2 specifically demonstrated superior performance in detecting genes and alleles compared to assembly-based methods in its original validation [25].
For traditional MLST schemes, stringMLST achieves 100% accuracy in less than 10 seconds per isolate according to some reports [23]. However, its performance may degrade with larger cgMLST schemes containing thousands of genes, where tools like MentaLiST (a successor to stringMLST) show superior scalability while maintaining accuracy [20].
When evaluating the capability to identify both correct alleles and new alleles, a 2019 study comparing SRST2, stringMLST, and STRAIN on 540 samples found varying performance levels. SRST2 demonstrated approximately 90% accuracy for correct allele identification, while stringMLST achieved slightly lower accuracy at 85-90% for traditional schemes [27]. ARIBA's local assembly approach provides advantages in identifying structural variations and gene disruptions, offering functional insights beyond mere sequence presence [26].
Computational Performance and Resource Requirements
Computational efficiency varies substantially between the three tools, reflecting their different algorithmic approaches:
Table 2: Computational Performance Comparison
| Tool | Processing Speed | Memory Usage | Scalability to cgMLST | Ease of Installation |
|---|---|---|---|---|
| SRST2 | Moderate (minutes per sample) | Moderate | Limited due to mapping overhead | Moderate (multiple dependencies) |
| ARIBA | Slower due to assembly step | Higher due to assembly | Moderate | Complex (multiple dependencies) |
| stringMLST | Very fast (seconds for traditional MLST) | Low | Limited for large schemes | Straightforward |
stringMLST typically demonstrates the fastest processing times for traditional MLST schemes, often completing typing in under 10 seconds per sample due to its efficient k-mer counting approach [23]. SRST2 requires moderate processing time (minutes per sample) due to the read mapping and statistical analysis steps [25]. ARIBA generally has the longest runtime due to its computationally intensive local assembly process [22] [26].
In terms of memory usage, stringMLST is the most efficient, followed by SRST2, while ARIBA typically requires the most memory due to its assembly component [22] [23]. For large-scale studies involving hundreds or thousands of isolates, these differences in computational requirements can significantly impact workflow feasibility.
Robustness to Challenging Sequencing Conditions
The performance of these tools under suboptimal sequencing conditions represents a crucial practical consideration for real-world applications:
Depth of Coverage: SRST2 has demonstrated reliable performance at coverages as low as 30x, though accuracy decreases substantially below this threshold [25]. stringMLST maintains accuracy down to approximately 20x coverage for traditional MLST schemes [23]. ARIBA's assembly-based approach requires higher coverage (typically >50x) for optimal performance, as low coverage can result in fragmented assemblies [26].
Sequence Contamination and Mixed Samples: SRST2 includes mechanisms to detect and flag mixed infections or contaminated samples by identifying multiple alleles at individual loci [25]. ARIBA's assembly approach can potentially separate contaminating sequences through its clustering algorithm [22]. stringMLST may struggle with mixed samples as its k-mer voting system assumes a pure isolate [23].
Novel Allele Detection: SRST2 flags potential novel alleles when exact matches are not found and can generate consensus sequences for further investigation [25]. ARIBA provides detailed information about variations from reference sequences, facilitating novel allele identification [26]. stringMLST has limited capability for novel allele characterization compared to the other tools [27].
Experimental Design and Implementation
Benchmarking Protocols
Robust evaluation of MLST typing tools requires carefully designed benchmarking approaches that assess performance across diverse conditions:
Reference Dataset Validation: Studies typically employ datasets with known sequence types determined by conventional methods. For example, one comprehensive comparison used datasets from the Gen-FS WGS Standards and Analysis Working Group, including C. jejuni, E. coli, L. monocytogenes, and S. enterica [22]. The validation of SRST2 utilized over 900 genomes from common pathogens with known MLST types [25].
Simulated Data Analysis: To systematically evaluate tool performance under controlled conditions, researchers often employ simulated reads with varying coverage depths (e.g., from 10x to 100x) and known contamination levels [22]. This approach allows for precise assessment of accuracy limits and failure modes.
Computational Resource Profiling: Benchmarking studies typically execute tools on standardized computing infrastructure while monitoring runtime, peak memory usage, and disk I/O through multiple iterations to ensure reproducible performance measurements [23] [20].
Table 3: Key Research Reagents and Computational Resources for MLST Analysis
| Resource Type | Specific Examples | Function in MLST Analysis |
|---|---|---|
| Reference Databases | PubMLST, BIGSdb, CARD, EnteroBase | Provide curated allele sequences and ST profiles for accurate typing |
| Sequencing Platforms | Illumina HiSeq/MiSeq, Nanopore | Generate raw sequencing data for input to typing tools |
| Alignment Tools | Bowtie2, Minimap, BWA | Perform read alignment for mapping-based approaches (SRST2, ARIBA) |
| Assembly Algorithms | SPAdes, Velvet, Fermi-lite | Reconstruct contiguous sequences from reads (ARIBA) |
| k-mer Counters | KAnalyze, Jellyfish | Index and count k-mers for k-mer-based approaches (stringMLST) |
| Programming Environments | Python, R, Julia, Perl | Provide execution environments for analysis tools and scripts |
Discussion and Practical Recommendations
Within the broader context of comparing CGF (cgMLST) versus traditional MLST for bacterial subtyping research, the selection of an appropriate typing tool depends on several factors, including the research question, scale of the study, available computational resources, and required resolution.
For traditional MLST schemes (6-8 genes) where speed is prioritized, stringMLST provides the fastest processing time with minimal computational resources, making it suitable for high-throughput screening of large isolate collections [23]. However, researchers should be aware of its limitations in detecting novel alleles and scaling to larger schemes.
For studies requiring comprehensive gene detection with functional interpretation, ARIBA offers advantages through its local assembly approach, which provides information about gene completeness and disruption [26]. This makes it particularly valuable for antimicrobial resistance studies where gene integrity correlates with phenotype.
For balanced performance across accuracy, novel allele detection, and reasonable computational requirements, SRST2 remains a robust choice, particularly for clinical and public health laboratories [25]. Its mapping-based approach provides reliable typing while flagging potential novel variants for further investigation.
For large-scale cgMLST schemes involving hundreds to thousands of genes, next-generation tools like MentaLiST and STing may be more appropriate than the three tools reviewed here, as they implement optimized k-mer algorithms specifically designed for scalability [23] [20]. These tools represent the evolving landscape of in silico typing methods that can keep pace with expanding genome-scale typing schemes.
As the field moves toward core genome and whole genome MLST approaches, computational efficiency and scalability become increasingly critical. The methodological differences between mapping-based, assembly-based, and k-mer-based approaches will continue to influence tool selection as typing schemes expand in size and complexity. Researchers should consider both their immediate typing needs and future directions when selecting tools for bacterial subtyping workflows.
Molecular typing methods are fundamental to bacterial subtyping for epidemiological surveillance, outbreak detection, and source attribution. The selection of an appropriate method balances discriminatory power, reproducibility, cost, and throughput. This guide objectively compares two established approaches: Comparative Genomic Fingerprinting (CGF) and Multi-Locus Sequence Typing (MLST), framing them within a broader thesis on their comparative performance for bacterial subtyping research. We detail the experimental workflows from raw sequencing data to final typings, supported by performance data and protocol details to inform researchers, scientists, and drug development professionals.
The evolution from traditional methods like pulsed-field gel electrophoresis (PFGE) towards sequence-based techniques has marked a paradigm shift in molecular epidemiology. MLST has provided a portable, reproducible system based on the sequences of internal fragments of housekeeping genes. In contrast, CGF leverages the presence or absence of accessory genes to generate highly discriminatory genetic fingerprints, offering a potentially more deployable solution for large-scale surveillance [28] [29] [5].
Methodological Principles and Workflows
Multi-Locus Sequence Typing (MLST)
Principle: MLST is a nucleotide sequence-based approach that characterizes bacterial isolates using the sequences of internal fragments of typically seven housekeeping genes. Each unique sequence for a gene is assigned an allele number, and the combination of alleles across all loci defines the Sequence Type (ST), providing an unambiguous profile for each isolate [30].
Workflow: The standard MLST workflow can be applied to both assembled genomes and raw sequencing reads.
- Input: The process begins with either assembled draft genomes/contigs in FASTA format or raw sequencing reads in FASTQ format. For paired-end reads, two files per sample are required, distinguished by patterns (e.g.,
_1and_2) [30]. - Processing: For raw reads, the first step involves quality control, including trimming of low-quality bases and adapter sequences. The target loci are then identified within the data.
- Alignment & Typing: The sequences of the housekeeping genes are extracted and aligned against a species-specific scheme of known alleles from a database like PubMLST. For each locus, the algorithm determines the best-matching allele based on identity and coverage [30].
- Result Interpretation: The output specifies the Sequence Type and flags any discrepancies:
- Matched: Complete match with all alleles (100% identity and coverage).
- Partial: Potential errors or novel SNPs detected (average identity/coverage ≤99%).
- Not Matched: No match found, often due to an incorrect MLST scheme selection [30].
Comparative Genomic Fingerprinting (CGF)
Principle: CGF is a multiplex PCR-based method that detects the presence or absence of a carefully selected set of accessory genes. These genes, identified through comparative genomic analysis as having high intraspecies variability, create a unique binary fingerprint for each strain. This method focuses on the accessory genome, which can provide higher resolution than methods targeting only core genes [28] [5].
Workflow: The development and application of CGF involve a structured process.
- Assay Development: This foundational phase involves whole-genome sequencing of diverse reference strains. Comparative genomic analysis identifies accessory genes with variable presence across the population. A final set of targets (e.g., 40 genes for CGF40) is selected through optimization to ensure high discrimination and concordance with a reference phylogeny [5].
- Sample Processing: DNA is extracted from bacterial isolates.
- Multiplex PCR: A multiplex PCR reaction is performed targeting the predefined set of accessory genes.
- Detection & Profiling: The presence or absence of each amplicon is detected, typically using capillary electrophoresis or microarrays, to generate a binary profile [28] [5].
- Cluster Analysis: Profiles are compared, and isolates are grouped into clades based on profile similarity (e.g., ≥90%) for epidemiological interpretation [5].
The following diagram illustrates the core pathways from raw data to final output for both methods, highlighting their parallel yet distinct processes.
Performance Comparison and Experimental Data
The choice between CGF and MLST involves trade-offs between resolution, throughput, and cost. The table below summarizes their performance characteristics based on experimental data.
Table 1: Comparative Performance of CGF and MLST for Bacterial Subtyping
| Feature | Comparative Genomic Fingerprinting (CGF) | Multi-Locus Sequence Typing (MLST) |
|---|---|---|
| Typing Principle | Presence/absence of accessory genes [28] [5] | Allelic profile of 7-8 housekeeping genes [29] [30] |
| Discriminatory Power | High; can differentiate closely related strains with distinct epidemiology [28] [5] | Medium; may lack resolution within common Sequence Types [29] |
| Throughput | High; suitable for large-scale surveillance [5] | Medium; more resource-intensive and lower throughput than CGF [5] |
| Reproducibility | High (98.6% reproducibility demonstrated) [5] | Very high; unambiguous sequence-based data [29] |
| Cost & Deployment | Lower cost; highly deployable for routine use [5] | Higher cost; can be cost-prohibitive for large studies [29] [5] |
| Data Portability | Binary profile; requires standardized gene set | Excellent; standardized, portable STs via curated databases (e.g., PubMLST) [28] [29] |
| Epidemiological Concordance | High concordance with outbreaks; identifies relevant clusters [28] | Good for long-term epidemiology; may miss recent outbreaks [28] |
| Representative Performance Metric | Simpson's Index of Diversity (ID) > 0.969 for CGF40 assay on A. butzleri [5] | Found 25% of isolates part of clusters in a sentinel surveillance study [28] |
Experimental data directly comparing these methods highlights their respective strengths. In a study on Campylobacter, CGF was identified as one of the optimal methods for detecting epidemiologically relevant clusters of cases. It could be effectively supplemented by flaA SVR sequencing, with or without MLST. The study concluded that different methods are optimal for uncovering different aspects of source attribution, and using multiple methods reveals more about a population than any single method alone [28].
Detailed Experimental Protocols
Protocol: MLST from Raw Reads
This protocol is adapted from standard procedures used in public health and research laboratories [28] [30].
- DNA Extraction & Input Preparation: Extract genomic DNA from bacterial isolates. The input for an MLST pipeline can be either:
- Raw Sequencing Reads: in FASTQ format (single or paired-end).
- Assembled Draft Genome: in FASTA format.
- Quality Control (for Raw Reads): Process raw reads using tools like Trimmomatic to remove low-quality bases, adapter sequences, and short reads. This step is crucial for ensuring accurate sequence alignment [31] [30].
- Scheme Selection: Select the appropriate species-specific MLST scheme from a curated database (e.g., PubMLST). Using an incorrect scheme will result in failed typing [30].
- Locus Identification & Allele Calling: The analysis tool (e.g., a dedicated MLST software) maps the reads or assembled contigs to the reference alleles for the seven housekeeping genes. For each locus, it determines the best-matching allele based on percentage identity and coverage [30].
- Sequence Type Assignment: The combination of the seven allele numbers defines the Sequence Type (ST). This ST is queried against the database to determine if it is a known or novel type. Results are often categorized as "Matched," "Partial" (indicating potential novel SNPs), or "Not Matched" [30].
Protocol: CGF Assay Implementation
This protocol is based on the development and application of CGF for pathogens like Campylobacter jejuni and Arcobacter butzleri [28] [5].
- CGF Assay Selection: Utilize a pre-defined set of accessory gene targets validated for the specific bacterial species. For example, the CGF40 assay for A. butzleri uses 40 genes selected for high discrimination and concordance with a reference phylogeny [5].
- DNA Extraction: Extract high-quality genomic DNA from bacterial isolates.
- Multiplex PCR: Perform a single, optimized multiplex PCR reaction that amplifies all target accessory genes simultaneously.
- Amplicon Detection & Scoring: Separate and detect the PCR amplicons using a platform such as capillary electrophoresis. Score each gene in the assay as "present" (1) or "absent" (0) based on the detection of its respective amplicon.
- Profile Analysis & Clustering: Analyze the binary profile of the isolate. Use a similarity coefficient (e.g, Jaccard) and cluster analysis (e.g., UPGMA) to group isolates into clades. A similarity threshold of ≥90% is often used to define clusters of genetically related isolates for epidemiological investigations [5].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of these typing workflows requires specific laboratory and bioinformatics reagents.
Table 2: Key Research Reagents and Solutions for Typing Workflows
| Item | Function in Workflow | Application |
|---|---|---|
| PureGene DNA Purification Kit | Genomic DNA purification from bacterial isolates, providing high-quality template for PCR or sequencing [28]. | CGF & MLST |
| PubMLST Database | Centralized repository for MLST schemes, allele sequences, and Sequence Type profiles, ensuring standardization and portability [28] [30]. | MLST |
| Trimmomatic | Bioinformatics tool for pre-processing raw FASTQ files; removes adapter sequences and trims low-quality bases to improve downstream analysis [31]. | MLST |
| Medifuge MF200 Centrifuge | Specialized centrifuge used in the preparation of samples, such as for concentrating growth factors or bacterial cells, prior to DNA extraction [32]. | Sample Prep |
| Bowtie 2 / Kraken 2 | Bioinformatics tools for read alignment and taxonomic classification, useful for filtering out contaminating reads from samples before targeted assembly or analysis [31]. | MLST |
| CGF Optimizer Software | Bioinformatic tool used to select an optimal subset of accessory genes for a CGF assay that maintains high concordance with a reference phylogeny [5]. | CGF |
| ChromatoGate | Open-source software for semi-automatic inspection of chromatograms from Sanger sequencing, aiding in the detection and correction of base mis-calls to ensure sequence accuracy [33]. | MLST (Sanger) |
Both CGF and MLST offer robust pathways from raw sequencing data to a definitive strain type or fingerprint, yet they serve complementary roles in the molecular epidemiologist's toolkit. MLST provides a standardized, portable, and phylogenetically meaningful framework ideal for global surveillance and population biology studies. In contrast, CGF offers a higher-resolution, high-throughput, and cost-effective alternative that is exceptionally well-suited for rapid outbreak detection and source tracking where fine-scale discrimination is required.
The decision between them should be guided by the specific research question, available resources, and desired balance between portability and discriminatory power. As whole-genome sequencing becomes increasingly accessible, methods like core-genome MLST (cgMLST) are emerging as new gold standards [29]. However, until WGS is universally deployable, CGF and MLST remain vital and highly effective methods for bacterial subtyping.
Sentinel surveillance systems are a cornerstone of public health, serving as an early-warning mechanism to detect clusters of infectious diseases before they become widespread outbreaks. For bacterial pathogens like Campylobacter and Salmonella, the effectiveness of these systems hinges on the resolution and speed of the molecular subtyping methods used to distinguish between strains [ [28]]. This guide provides a comparative analysis of two prominent subtyping methods—Comparative Genomic Fingerprinting (CGF) and Multilocus Sequence Typing (MLST)—evaluating their performance, protocols, and applicability within modern sentinel surveillance frameworks.
To understand their comparative performance, it is essential to first define the fundamental principles and technical execution of each method.
Multilocus Sequence Typing (MLST)
MLST is a gold-standard, sequence-based technique that characterizes bacterial isolates by sequencing approximately 450-500 bp internal fragments of seven housekeeping genes. The sequences for each locus are assigned as distinct alleles, and the combination of alleles across the seven genes defines the sequence type (ST) of the isolate. This method is highly reproducible and portable, making it excellent for long-term, global epidemiological studies and population genetics [ [4] [28]].
Workflow Diagram: MLST
Comparative Genomic Fingerprinting (CGF)
CGF is a higher-resolution, PCR-based method that targets genomic variation within the accessory genome. Instead of sequencing, it detects the presence or absence of multiple, highly variable marker genes distributed across the genome. The CGF40 assay, for example, uses a 40-gene multiplex PCR panel to generate a binary fingerprint for each isolate. This fingerprint reflects strain-specific genetic content and has been shown to provide greater discriminatory power than MLST [ [4]].
Workflow Diagram: CGF
Head-to-Head Performance Comparison
The core of this guide lies in the direct, data-driven comparison of CGF and MLST, focusing on metrics critical for sentinel surveillance.
Key Performance Metrics for Sentinel Surveillance
The table below summarizes experimental data from a validation study of 412 C. jejuni isolates from various sources, directly comparing CGF40 and MLST [ [4]].
Table 1: Quantitative Performance Comparison of CGF40 and MLST
| Feature | MLST (Sequence Type) | MLST (Clonal Complex) | CGF40 |
|---|---|---|---|
| Simpson's Index of Diversity (ID) | 0.935 | 0.873 | 0.994 |
| Primary Target | Core genome (housekeeping genes) | Core genome (housekeeping genes) | Accessory genome (variable genes) |
| Methodology | Sequencing & allele assignment | Sequencing & clonal complex assignment | Multiplex PCR & presence/absence profiling |
| Discriminatory Power | High | Moderate | Very High |
| Cost & Speed | Higher cost, slower turnaround | Higher cost, slower turnaround | Lower cost, rapid results |
| Best Application | Long-term epidemiology, population structure | Long-term epidemiology, population structure | Short-term outbreak detection, cluster investigation |
Interpretation of Comparative Data
- Discriminatory Power: The significantly higher Simpson's Index of Diversity for CGF40 (0.994) demonstrates its superior ability to differentiate between closely related bacterial isolates. This is crucial in sentinel surveillance for distinguishing outbreak clusters from a background of sporadic cases, particularly for common sequence types like ST21 and ST45 of C. jejuni [ [4]].
- Epidemiological Concordance: Despite its higher resolution, CGF maintains high concordance with MLST. Isolates that are identical by MLST often resolve into distinct but highly similar CGF profiles, confirming their relatedness while revealing finer-scale transmission patterns [ [4]].
- Operational Utility: CGF is noted for being rapid, lower in cost, and more easily deployable for routine surveillance compared to the more laborious and expensive sequencing required for MLST [ [4] [28]].
Experimental Protocols for Validation
For researchers seeking to validate or implement these methods, the following summarized protocols are essential.
Detailed CGF40 Assay Protocol
The CGF40 method for C. jejuni, as described by Taboada et al., can be broken down into key stages [ [4]]:
- Marker Selection and Assay Design: Forty target genes were selected based on five criteria:
- Documented absence in one or more strains from prior genomic surveys.
- Unbiased carriage across population datasets (avoiding universally present/absent genes).
- Representative genomic distribution across 16 major hypervariable regions.
- Ability to recapitulate strain relationships from whole-genome analysis.
- Presence in multiple reference genomes to allow for SNP-free PCR primer design.
- Primer Design and Multiplexing: SNP-free PCR primers were designed for each of the 40 targets. These were assembled into 8 multiplex PCRs, each targeting 5 distinct loci.
- Wet-Lab Procedure:
- DNA Extraction: Use a commercial kit (e.g., PureGene) for high-quality genomic DNA.
- Multiplex PCR: Perform the 8 parallel PCR reactions under optimized conditions.
- Data Generation: Analyze PCR products to generate a binary presence/absence profile for all 40 loci.
- Data Analysis and Fingerprinting: The combined profile is used to generate a comparative genomic fingerprint for cluster analysis.
Standard MLST Protocol
The standard MLST protocol for C. jejuni, as referenced in the studies, follows these steps [ [4] [28]]:
- DNA Extraction: Use a standardized method (e.g., PureGene kit) for genomic DNA preparation.
- PCR Amplification: Independently amplify the seven housekeeping genes (aspA, glnA, gltA, glyA, pgm, tkt, uncA).
- Sequencing and Assembly: Purify PCR amplicons (e.g., using Montage PCR centrifugal filters) and perform Sanger sequencing. Assemble and check sequences for quality using software like SeqMan.
- Allele and ST Assignment: Query the curated Campylobacter PubMLST database (http://pubmlst.org/campylobacter/) to assign allele numbers and determine the final Sequence Type.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Materials for CGF and MLST Protocols
| Reagent / Material | Function in Protocol | Example Product / Note |
|---|---|---|
| Genomic DNA Purification Kit | Isolation of high-quality, PCR-ready genomic DNA from bacterial isolates. | PureGene Genomic DNA Purification Kit (Gentra Systems) [ [4]] |
| PCR Primers | Specific amplification of target loci (7 for MLST, 40 for CGF). | Custom-designed primers; for CGF, assembled into multiplex reactions [ [4]] |
| PCR Purification Kit | Post-amplification cleanup of PCR products prior to sequencing (for MLST). | Montage PCR Centrifugal Filter Devices [ [4]] |
| Cycle Sequencing Kit | Sanger sequencing of amplified gene fragments (for MLST). | BigDye Terminator 3.1 Chemistry (Applied Biosystems) [ [4]] |
| Capillary Electrophoresis System | Separation and detection of sequenced fragments (for MLST). | ABI 3100 or 3730 DNA Analyzer (Applied Biosystems) [ [4]] |
| Sequence Assembly & Analysis Software | Assembly of sequencing reads, quality control, and allele calling. | SeqMan (DNASTAR Lasergene suite) [ [4]] |
| Curated MLST Database | Centralized resource for allele and sequence type assignment. | PubMLST (http://pubmlst.org/campylobacter/) [ [28]] |
The choice between CGF and MLST for sentinel surveillance is not a matter of identifying a single superior technique, but of selecting the right tool for the specific public health question. CGF offers a powerful, high-resolution, and cost-effective solution for the real-time detection and investigation of disease clusters, making it highly suitable for routine surveillance and outbreak management. In contrast, MLST remains the definitive method for understanding long-term, global population structures and evolutionary relationships.
The field continues to evolve, with Whole Genome Sequencing (WGS)-based methods like core genome MLST (cgMLST) emerging as powerful successors that offer ultimate resolution and standardization [ [34]]. However, for many laboratories, the balance of speed, cost, and discriminatory power ensures that CGF remains a highly relevant and effective tool for protecting public health through robust sentinel surveillance.
Campylobacter jejuni and C. coli are the most common bacterial causes of gastroenteritis worldwide, representing a significant public health and socioeconomic burden [35] [36]. Despite its high incidence, tracking the sources of sporadic campylobacteriosis remains challenging, primarily due to the limitations of existing molecular typing methods in unambiguously linking genetically related strains [35]. The genomic evolution of Campylobacter is characterized by frequent rearrangements and interstrain genetic exchange, which complicates the interpretation of molecular typing data for outbreak investigations [35] [6].
Multiple molecular subtyping methods have been developed for Campylobacter, including multi-locus sequence typing (MLST), flagellin gene typing (flaA-SVR), porA gene typing, and pulsed-field gel electrophoresis (PFGE) [35] [28]. While these methods have advanced our understanding of Campylobacter epidemiology, they present limitations for routine surveillance and outbreak detection, including insufficient discriminatory power, high costs, technical complexity, and prolonged turnaround times [37] [28].
This case study examines Comparative Genomic Fingerprinting (CGF) as an optimal method for Campylobacter outbreak detection. We evaluate its performance against established typing methods, with a particular focus on its application in public health surveillance and epidemiological investigations.
Understanding the Typing Landscape for Campylobacter
Established Typing Methods and Their Limitations
Multi-locus sequence typing (MLST), which analyzes DNA sequences of seven housekeeping genes, has become a leading method for Campylobacter subtyping due to its portability and ease of interlaboratory comparison [35] [4]. However, MLST may lack sufficient resolution for short-term investigations aimed at identifying temporally and spatially related clusters from common sources [4]. Additionally, MLST is resource-intensive and relatively low-throughput, limiting the number of isolates that can be analyzed in most laboratory settings [5].
Single-locus methods such as flaA-SVR and porA typing offer simpler alternatives but provide less discriminatory power than multi-locus approaches [35] [28]. Pulsed-field gel electrophoresis (PFGE) has been valuable for outbreak investigations but is of limited value for Campylobacter due to chromosomal rearrangements and high genetic diversity that may limit the clustering of related isolates [4].
The Emergence of Comparative Genomic Fingerprinting (CGF)
Comparative Genomic Fingerprinting was developed to overcome the technical and logistical hurdles of implementing Campylobacter typing in routine surveillance [37] [4]. This method uses a multiplex PCR approach to detect the presence or absence of multiple genes in the accessory genome, creating a binary fingerprint that distinguishes strains based on differences in genome content [4].
The CGF40 assay, which targets 40 accessory genes, was specifically designed as a rapid, low-cost, and high-resolution subtyping method suitable for large-scale epidemiological surveillance [4] [38]. The selection of target genes was based on comprehensive genomic analyses, choosing markers with adequate carriage across populations and a representative genomic distribution to ensure optimal discrimination of strains [4].
Table 1: Key Characteristics of Major Campylobacter Subtyping Methods
| Method | Target | Discriminatory Power | Throughput | Cost | Technical Demand |
|---|---|---|---|---|---|
| CGF40 | Accessory genome (40 genes) | High (ID: 0.994) [4] | High | Low | Moderate |
| MLST | Core genome (7 housekeeping genes) | Moderate (ID: 0.935) [4] | Low | High | High |
| flaA-SVR | Single locus (flagellin gene) | Low [35] | Moderate | Moderate | Moderate |
| porA | Single locus (porin gene) | Low [35] | Moderate | Moderate | Moderate |
| PFGE | Whole genome macrorestriction | Variable [4] | Low | Moderate | High |
Comparative Performance: CGF Versus MLST
Discriminatory Power and Resolution
Multiple studies have demonstrated that CGF40 provides superior discriminatory power compared to MLST. In a comprehensive validation study analyzing 412 C. jejuni isolates from various sources, CGF40 exhibited a Simpson's index of diversity (ID) of 0.994, significantly higher than MLST at both the sequence type (ST) level (ID = 0.935) and clonal complex (CC) level (ID = 0.873) [4].
This enhanced resolution is particularly valuable for differentiating closely related isolates within prevalent sequence types. CGF has been shown to effectively discriminate between isolates with identical MLST profiles, partitioning them into distinct but highly similar CGF profiles that may reflect epidemiological differences [4] [38]. This capability is crucial for outbreak detection, where slight genetic variations between isolates must be identified to trace transmission pathways accurately.
Concordance with Evolutionary Relationships
Despite targeting different genomic elements (accessory genes versus core genes), CGF and MLST show high concordance in their grouping of isolates. High Wallace coefficients obtained when CGF40 was used as the primary typing method confirm this strong agreement between the two methods [4].
When evaluated against a "gold standard" reference phylogeny based on highly conserved core genes, both MLST and CGF provided better estimates of true phylogenetic relationships than single-locus methods (porA, flaA) [35]. This suggests that both multi-locus methods, despite their different targets, capture essential aspects of strain relationships relevant to epidemiological investigations.
Table 2: Performance Metrics of CGF40 Versus MLST for C. jejuni Subtyping
| Performance Metric | CGF40 | MLST (Sequence Type) | Reference |
|---|---|---|---|
| Simpson's Index of Diversity | 0.994 | 0.935 | [4] |
| Concordance with Reference Phylogeny | High (Multi-locus method) | High (Multi-locus method) | [35] |
| Cost per Isolate | ~$20 (Canadian) | ~$100 (Canadian) | [39] |
| Laboratory Time | 35 hours for 84 isolates | Significantly higher | [39] |
| Ease of Interlaboratory Comparison | High | High | [35] [4] |
CGF in Public Health Surveillance and Outbreak Detection
Enhanced Cluster Detection in Routine Surveillance
The implementation of CGF40 in public health surveillance has demonstrated its practical value for detecting clusters of campylobacteriosis that might otherwise go unrecognized. A prospective study in Nova Scotia, Canada, linked epidemiological data with CGF40 subtyping results for 299 cases reported between 2012 and 2015 [37].
The study identified 141 distinct CGF40 subtypes among the cases, with 70% of isolates sharing fingerprints with one or more isolates, suggesting possible common sources [37]. CGF40 successfully discerned known epidemiologically related isolates and augmented case-finding efforts, confirming its epidemiological validity [37].
Identification of Subtype-Specific Risk Factors
The application of CGF40 in surveillance enabled a case-case study design to examine risk factors for the most common CGF40 subtypes [37]. This approach revealed statistically significant associations between specific subtypes and particular exposure risks:
- Rural residence was associated with certain subtypes
- Local exposure patterns varied by subtype
- Contact with pet dogs or cats was linked to specific subtypes
- Contact with chickens showed subtype-specific associations
- Drinking unpasteurized milk was a risk factor for particular subtypes [37]
These findings demonstrate how CGF subtyping can elucidate the epidemiology of campylobacteriosis with greater precision than species-level identification alone, providing a starting point for outbreak hypothesis generation for specific CGF40 subtypes [37].
Methodological Protocols
CGF40 Assay Workflow
The CGF40 method employs eight multiplex PCR reactions, each targeting five loci, for a total of 40 accessory genes [4]. The assay workflow consists of:
- DNA extraction from Campylobacter isolates using standard genomic DNA purification methods
- Multiplex PCR amplification using eight pre-optimized primer sets
- PCR product analysis by gel electrophoresis or capillary electrophoresis
- Binary scoring of each target amplicon as present (1) or absent (0)
- Profile generation to create a unique CGF40 fingerprint for each isolate
- Subtype assignment by comparison with a reference database [4]
Experimental Validation Framework
The validation of CGF against other typing methods has been facilitated by the development of computational frameworks that use whole genome sequence (WGS) data as a "gold standard" [35] [6]. This approach involves:
- Whole genome sequencing of Campylobacter isolates
- In silico determination of multiple molecular types (MLST, flaA, porA, CGF)
- Reference phylogeny inference based on highly conserved core genes
- Concordance analysis using statistical measures such as the adjusted Wallace coefficient (AWC) [35]
This framework allows for rigorous assessment of typing method performance against the reference standard of whole genome relationships, providing objective metrics for method comparison [35] [6].
Essential Research Reagents and Tools
Table 3: Research Reagent Solutions for CGF Implementation
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| Genomic DNA Purification Kit | Template DNA preparation | Standard commercial kits sufficient [4] |
| CGF40 Primer Sets | Amplification of 40 target genes | Eight multiplex sets, five primers each [4] |
| Multiplex PCR Master Mix | Simultaneous amplification of multiple targets | Must maintain efficiency with multiple primers [4] |
| Gel Electrophoresis System | PCR product separation and visualization | Alternative: capillary electrophoresis for higher throughput [4] |
| CGF Reference Database | Subtype assignment and cluster analysis | Contains CGF40 profiles from diverse sources [37] |
Comparative Genomic Fingerprinting represents an optimal balance of resolution, throughput, and cost-effectiveness for Campylobacter outbreak detection and surveillance. The method's superior discriminatory power compared to MLST, combined with its technical accessibility and rapid turnaround time, makes it particularly suitable for public health laboratories tasked with monitoring and investigating campylobacteriosis.
The strong epidemiological validity demonstrated through prospective surveillance studies, coupled with the ability to identify subtype-specific risk factors, positions CGF as a valuable tool for advancing our understanding of Campylobacter transmission dynamics. While whole genome sequencing may eventually become the standard for microbial subtyping, CGF provides an effective solution for the current needs of public health surveillance and outbreak response.
For researchers and public health professionals, CGF offers a practical pathway to enhanced Campylobacter surveillance that can detect outbreaks more effectively than traditional methods while providing actionable insights for targeted intervention strategies.
In the field of epidemiology, source attribution refers to a category of methods with the objective of reconstructing the transmission of an infectious disease from a specific source, such as a population, individual, or location [40]. For foodborne pathogens like Campylobacter jejuni and C. coli, accurately tracing transmission pathways remains challenging due to their widespread distribution in animal and environmental reservoirs [28] [35]. Molecular source attribution uses the genetic characteristics of pathogens—most often their nucleic acid genome—to reconstruct transmission events with greater precision than traditional methods [40]. The fundamental assumption underlying these methods is that pathogens undergo minimal genetic change when transmitted between hosts, meaning that infections with genetically similar pathogens are likely to be epidemiologically related [40].
The evolution of typing technologies has progressed from phenotypic methods and serotyping to molecular techniques including pulsed-field gel electrophoresis (PFGE), and now to sequence-based methods such as multi-locus sequence typing (MLST) and whole-genome sequencing (WGS) [41]. Among current methodologies, Comparative Genomic Fingerprinting (CGF) and Multilocus Sequence Typing (MLST) have emerged as important tools for bacterial subtyping in public health surveillance and outbreak investigations [28] [42]. This guide provides a comparative analysis of these methods, focusing on their performance characteristics, applications, and suitability for different research scenarios.
Multi-Locus Sequence Typing (MLST)
MLST is a sequence-based typing approach that involves the amplification and sequencing of approximately seven housekeeping genes to characterize bacterial isolates [41] [40]. These genes are selected for their indispensable biological functions and presence across all members of a species, making them stable targets for comparison [40]. Each unique sequence is assigned an allele number, and the combination of alleles across the seven loci defines the sequence type (ST) for each isolate [41] [35]. The standardized nature of MLST allows for easy comparison of data across laboratories through curated databases such as PubMLST [41] [40].
MLST detects changes at the DNA level that cannot be inferred from phenotypic methods, providing a higher resolution than traditional serotyping [41]. However, its reliance on a limited number of conserved genes can limit its discriminatory power for outbreak detection, as genetic variation in housekeeping genes may not accumulate rapidly enough to distinguish between closely related strains [41] [35]. This has led to the development of extended schemes such as core genome MLST (cgMLST) which expands the analysis to hundreds or thousands of gene loci distributed throughout the core genome [41].
Comparative Genomic Fingerprinting (CGF)
CGF represents a different approach that targets the accessory genome—genes that are variably present among strains within a species [28] [42]. This method was developed to provide high-resolution subtyping suitable for large-scale epidemiological surveillance [42]. The CGF method typically uses multiplex PCR to detect the presence or absence of a carefully selected set of accessory genes, generating a binary fingerprint for each isolate [28] [35].
CGF was originally developed for C. jejuni using genes identified through comparative genomic hybridization as having high intraspecies variability [28]. A similar approach has been successfully applied to other pathogens such as Arcobacter butzleri, where a 40-gene CGF assay (CGF40) demonstrated high discriminatory power with a Simpson's Index of Diversity greater than 0.969 [42]. Unlike sequence-based methods, CGF focuses on genomic insertions and deletions, which can provide different insights into strain relationships and evolutionary history [35].
Whole Genome Sequencing (WGS) Based Approaches
While not the primary focus of this comparison, it is important to acknowledge that whole genome sequencing (WGS) is increasingly becoming the gold standard for bacterial subtyping [41] [35]. WGS-based methods include core genome MLST (cgMLST), whole genome MLST (wgMLST), and single nucleotide polymorphism (SNP)-based analysis [41]. These approaches provide the highest possible resolution for distinguishing bacterial strains but remain resource-intensive in terms of cost, computational requirements, and bioinformatics expertise [42] [41] [35]. As such, methods like CGF and MLST continue to play important roles in surveillance and epidemiology, particularly in settings where WGS is not yet feasible for routine use [42].
Table 1: Key Characteristics of Major Typing Methods
| Method | Genetic Target | Resolution | Throughput | Primary Application |
|---|---|---|---|---|
| MLST | Sequences of 7 housekeeping genes (core genome) | Moderate | Moderate | Long-term epidemiology, population structure studies |
| CGF | Presence/absence of 40+ accessory genes | High | High | Outbreak detection, cluster investigation, source tracking |
| cgMLST | Sequences of hundreds to thousands of core genes | Very High | Low (requires WGS) | High-resolution outbreak investigation, transmission tracing |
| SNP-based | Single nucleotide variants across entire genome | Highest | Low (requires WGS) | Precise transmission chain resolution, evolutionary studies |
Comparative Performance Analysis
Discriminatory Power and Resolution
Studies directly comparing CGF and MLST have demonstrated important differences in their ability to distinguish between closely related strains. In an analysis of 104 C. jejuni and C. coli genomes, both MLST and CGF provided better estimates of true phylogenetic relationships than single-locus methods (porA, flaA) when compared to a reference phylogeny based on 389 highly conserved core genes [35]. However, CGF generally exhibits higher discriminatory power than MLST, allowing differentiation of closely related strains with distinct epidemiology [42] [35].
The enhanced resolution of CGF is particularly valuable for detecting clusters of cases that may represent outbreaks. Research within the C-EnterNet sentinel site surveillance program in Canada identified CGF as the optimal method for detecting epidemiologically relevant clusters of Campylobacter isolates, potentially indicating outbreaks [28]. The method's focus on the accessory genome, which may evolve more rapidly than the core genome targeted by MLST, likely contributes to this improved discriminatory power [35].
Epidemiological Concordance and Cluster Detection
The ability of a typing method to accurately reflect epidemiological relationships between isolates is crucial for effective public health response. Both MLST and CGF show high concordance with inferred transmission pathways, but they may excel in different scenarios. MLST, targeting stable housekeeping genes, provides a robust framework for understanding long-term population structure and evolutionary relationships [40] [35]. In contrast, CGF appears particularly well-suited for detecting recent transmission events and identifying clusters that might be missed by other methods [28].
In one sentinel site study, CGF proved optimal for detecting clusters of Campylobacter cases, while flaA SVR sequencing and MLST could identify additional clusters potentially linked to infections from multiple or different sources [28]. This suggests that a combined approach using multiple typing methods may provide the most comprehensive understanding of transmission dynamics, revealing more about the population structure than any single method alone [28].
Practical Implementation Considerations
From a practical standpoint, CGF and MLST differ significantly in their requirements for instrumentation, technical expertise, and data analysis. MLST relies on DNA sequencing of multiple loci, which can be resource-intensive and may limit throughput [42]. While MLST generates portable data that can be easily shared between laboratories through standardized databases, the requirement for sequencing can create bottlenecks in large-scale surveillance [42] [41].
CGF, implemented through multiplex PCR, offers higher throughput and lower per-sample cost compared to MLST, making it more suitable for routine surveillance activities [42]. The binary nature of CGF results (presence/absence of target genes) also simplifies data analysis compared to sequence-based methods [28] [42]. However, CGF requires careful validation and optimization of gene targets for different bacterial species, whereas MLST schemes are already established for many important pathogens [42].
Table 2: Performance Comparison of CGF vs. MLST for Campylobacter spp.
| Performance Metric | MLST | CGF | Experimental Context |
|---|---|---|---|
| Adjusted Wallace Coefficient (AWC) vs. reference phylogeny | 0.66 (0.54-0.78) | 0.65 (0.53-0.77) | Comparison with phylogeny of 389 highly conserved core genes from 104 C. jejuni and C. coli genomes [35] |
| Cluster Detection Capability | Moderate | High | Sentinel site surveillance; CGF optimal for detecting epidemiologically relevant clusters [28] |
| Discriminatory Power | Moderate | High | CGF showed improved discrimination of closely related strains with distinct epidemiology [42] [35] |
| Throughput | Moderate | High | CGF based on multiplex PCR more suitable for large-scale surveillance than sequence-based MLST [28] [42] |
| Cost and Deployment | Higher cost, requires sequencing | Lower cost, more deployable for routine surveillance | CGF provides practical alternative in resource-limited settings [42] |
Experimental Protocols and Methodologies
Standard MLST Protocol for Campylobacter spp.
The standard MLST protocol for Campylobacter jejuni and C. coli involves amplification and sequencing of seven housekeeping genes: aspA (aspartase A), glnA (glutamine synthetase), gltA (citrate synthase), glyA (serine hydroxymethyltransferase), pgm (phosphoglucomutase), tkt (transketolase), and uncA (ATP synthase alpha subunit) [28] [35].
Detailed Methodology:
- DNA Extraction: Genomic DNA is purified from bacterial cultures using commercial kits such as the PureGene genomic DNA purification kit [28].
- PCR Amplification: Each locus is amplified separately using sequence-specific primers under standardized cycling conditions [28] [35].
- Product Cleanup: PCR products are purified using centrifugal filter devices or enzymatic cleanup to remove primers and nucleotides [28].
- DNA Sequencing: Bidirectional sequencing is performed using the amplification primers or internal sequencing primers [28].
- Sequence Analysis: Sequences are trimmed and compared to existing alleles in the Campylobacter MLST database (http://pubmlst.org/campylobacter/) to assign allele numbers and sequence types [28] [35].
The entire process typically requires 2-3 days to complete, with sequencing being the rate-limiting step. Consistency in data interpretation is maintained through curated databases that map sequences to a fixed notation of allele designations [40].
CGF Methodology for Bacterial Subtyping
The CGF method uses a different approach, targeting presence or absence of accessory genes through multiplex PCR. For C. jejuni, the CGF method detects 40 genes that were identified as having a high degree of intraspecies variability in comparative genomic hybridizations using DNA microarrays [28] [35].
Detailed Methodology:
- Gene Selection: Comparative analysis of genome sequences identifies accessory genes suitable for generating unique genetic fingerprints based on gene presence/absence patterns [42].
- Primer Design: Primers are designed to amplify each of the target genes and organized into multiplex PCR panels [42].
- Multiplex PCR: DNA is amplified using optimized multiplex PCR conditions that allow simultaneous detection of multiple targets [28] [42].
- Product Detection: Amplification products are typically separated by capillary electrophoresis to determine which target genes are present [28] [42].
- Profile Generation: Results are compiled into a binary profile (1 for presence, 0 for absence) for each isolate, which can be compared to databases of known profiles [42].
The CGF40 assay for Arcobacter butzleri follows a similar workflow, employing a set of 40 accessory genes identified through comparative genomic analysis of diverse strains [42]. This method can be completed within 1-2 days, with higher throughput potential than sequence-based methods.
Workflow Comparison Diagram
Essential Research Reagents and Tools
Table 3: Essential Research Reagents for Molecular Typing Methods
| Reagent/Equipment | Function in Typing Workflow | Method Application |
|---|---|---|
| PureGene DNA Purification Kit (Gentra Systems) | Genomic DNA extraction from bacterial cultures | Used in both MLST and CGF protocols for consistent DNA quality [28] |
| Sequence-Specific Primers | Amplification of target genes (housekeeping or accessory) | MLST: 7 pairs of primers for housekeeping genes; CGF: Multiplex primer sets for accessory genes [28] [42] |
| Montage PCR Centrifugal Filter Devices | PCR product cleanup prior to sequencing | Essential for MLST to remove excess primers and nucleotides before sequencing [28] |
| Capillary Electrophoresis System | Separation and detection of multiplex PCR products | Used in CGF to determine presence/absence of target genes based on amplicon size [42] |
| Sanger Sequencing Platform | Determination of DNA sequence for target genes | Core component of MLST methodology for obtaining sequence data [28] [35] |
| Curated Reference Databases (e.g., PubMLST) | Assignment of allele numbers and sequence types | Essential for MLST data interpretation and standardization across laboratories [28] [41] [40] |
The comparative analysis of CGF and MLST reveals distinct strengths and optimal applications for each method in bacterial source attribution studies. CGF offers advantages in outbreak detection and cluster investigations due to its high discriminatory power, throughput, and practical deployability in routine surveillance [28] [42]. Its focus on the accessory genome provides resolution for distinguishing closely related strains that might appear identical by MLST [35]. Conversely, MLST remains valuable for understanding population structure, long-term epidemiology, and evolutionary relationships due to its standardized framework and curated databases [41] [40] [35].
The choice between these methods should be guided by specific research objectives, available resources, and the required balance between resolution and throughput. For comprehensive surveillance programs, a combined approach using CGF for initial cluster detection followed by MLST for broader contextualization may provide the most complete epidemiological picture [28]. As WGS technologies continue to become more accessible and affordable, they will likely supersede both methods for high-priority investigations [41] [35]. However, for the foreseeable future, CGF and MLST will remain important tools in the molecular epidemiology toolkit, particularly for large-scale surveillance and in resource-limited settings where WGS implementation remains challenging [42].
Navigating Challenges: From Mixed Samples to Computational Limits
Within bacterial subtyping for public health surveillance and outbreak detection, two primary genotyping methods are widely used: Comparative Genomic Fingerprinting (CGF) and Multilocus Sequence Typing (MLST). The performance of these methods under suboptimal conditions—specifically with mixed microbial samples or low sequencing coverage—represents a critical practical consideration for researchers and public health laboratories. This guide provides an objective comparison of CGF and MLST resilience, synthesizing experimental data from direct methodological comparisons to inform selection for routine surveillance and outbreak investigations.
Methodological Foundations
Core Principles and Workflows
Multilocus Sequence Typing (MLST) characterizes bacterial strains through the sequencing of approximately 450-500 bp internal fragments of seven housekeeping genes [43]. The sequences are compared to a central database to assign allele numbers and sequence types (STs), providing a standardized, portable typing system suitable for long-term and global epidemiology [4].
Comparative Genomic Fingerprinting (CGF) employs multiplex PCR to amplify multiple (e.g., 40) accessory genomic regions distributed throughout the genome, detecting presence or absence variations [4]. This method targets the accessory genome, capturing a different aspect of genetic variation compared to MLST's focus on housekeeping genes.
The fundamental workflows for these methods, particularly when derived from Whole Genome Sequencing (WGS) data, are illustrated below.
Research Reagent Solutions
The following table details essential reagents and materials required for implementing these typing methods, particularly in the context of WGS-based workflows.
| Reagent/Material | Function in Typing Workflow | Application in CGF vs. MLST |
|---|---|---|
| PureGene DNA Purification Kit (Gentra Systems) | Genomic DNA purification for downstream molecular analyses | Used in conventional MLST and CGF method development for template preparation [4] [28] |
| Kapa HyperPlus Library Prep Kit (Roche) | Library preparation for next-generation sequencing | Enables WGS data generation for both assembly- and mapping-based MLST derivation [44] |
| Montage PCR Centrifugal Filters (Fisher Scientific) | Purification of PCR amplicons prior to sequencing | Critical for cleaning up MLST PCR products for Sanger sequencing [4] |
| NovaSeq 6000 (Illumina) | High-throughput sequencing platform | Generates short-read data for WGS-based subtyping; coverage depth impacts MLST call reliability [43] [44] |
| Nanopore Flow Cells (Oxford Nanopore) | Long-read sequencing platform | Useful for resolving complex genomic regions and structural variants impacting accessory gene content [45] [46] |
Performance Comparison Under Challenging Conditions
Resilience to Low Sequencing Coverage
Low-coverage Whole Genome Sequencing (lcWGS) presents a significant challenge for sequence-based typing methods. Experimental data demonstrates that mapping-based MLST approaches show notable resilience in this context. One systematic evaluation found that with low-coverage samples (minimum read depth of 1-10×), a mapping-based method could derive full MLST profiles for 89.1% (49/55) of samples, outperforming assembly-based approaches which succeeded for only 67.3% (37/55) [43]. This highlights a key advantage of read-mapping strategies for maximizing information yield from limited sequencing data.
For CGF, which typically relies on predefined PCR amplification, low DNA yield or quality can impact results. However, optimized PCR-CGP (comprehensive genomic profiling) tests have demonstrated success even with suboptimal samples. One study reported that 80.5% of exception samples (those not meeting minimum input requirements for TC, TSA, or nucleic acid yield) still yielded reportable results [47], suggesting that targeted amplification approaches can maintain functionality when sequencing-based methods might fail.
Handling Mixed Microbial Samples
Mixed samples containing multiple bacterial species or strains present distinct challenges for subtyping methods. Experimental evidence indicates that mapping-based MLST maintains superior sensitivity in these scenarios. In samples containing Salmonella enterica mixed with other species like Proteus mirabilis or Escherichia coli, the mapping-based approach successfully derived sequence types for all tested mixtures, while assembly-based methods frequently returned "undetermined" results [43].
For CGF, which generates strain-specific fingerprints based on accessory gene content, mixed samples can complicate profile interpretation. However, CGF's targeting of multiple dispersed loci may potentially help distinguish co-occurring strains through differential amplification patterns, though specific quantitative data on CGF performance with defined mixed samples is less extensively documented than for MLST.
Quantitative Performance Comparison
The table below summarizes key performance metrics for CGF and MLST based on comparative studies.
| Performance Metric | CGF40 (C. jejuni) | MLST (C. jejuni) | Experimental Context |
|---|---|---|---|
| Simpson's Index of Diversity (ID) | 0.994 [4] | 0.935 (ST), 0.873 (CC) [4] | 412 isolates from multiple sources |
| Profile Success Rate (Low Coverage) | Information Limited | 89.1% (mapping), 67.3% (assembly) [43] | 55 mixed and low coverage genomes |
| Profile Success Rate (Mixed Samples) | Information Limited | 100% (mapping), Variable (assembly) [43] | 26 mixed genomic data sets |
| Concordance with Conventional Method | 100% [5] | 92.9% [43] | 323 bacterial genomes of diverse species |
| Typing Concordance (Wallace Coefficient) | High concordance with MLST [4] | High concordance with CGF [4] | 412 C. jejuni isolates |
Experimental Protocols for Method Evaluation
Protocol for Assessing MLST Performance with Mixed Samples
To evaluate MLST reliability with mixed samples, researchers have employed the following validated protocol:
Sample Preparation: Create intentional mixtures of genomic DNA from different bacterial strains or species at defined ratios (e.g., 90:10, 80:20, down to 50:50) [43]. Use DNA extraction methods such as the Qiasymphony (Qiagen) to ensure high-quality input material.
Whole Genome Sequencing: Sequence the mixed samples using platforms such as Illumina to generate short-read data. Standardize DNA quantification (e.g., using Glomax, Promega) and ensure mixed concentrations (e.g., 25 ng/μL in a final volume of 75 μL) [43].
Bioinformatic Analysis:
- Assembly-Based MLST: Perform de novo assembly of reads into contigs, then compare these contigs to a reference allele database using BLAST to assign MLST types [43].
- Mapping-Based MLST: Align reads directly to reference allele sequences using tools like BWA or Bowtie2, then call variants using algorithms such as Samtools mpileup to determine the most likely allele at each locus [43].
Quality Assessment: For mapping-based approaches, calculate quality metrics such as "maximum percentage non-consensus base values" to quantitatively assess mixture detection [43].
Protocol for Evaluating CGF Discriminatory Power
To validate CGF assay performance and compare it with MLST:
Strain Selection: Curate a diverse set of bacterial isolates from multiple sources (human clinical, agricultural, environmental, retail) to ensure representative sampling [4].
Parallel Typing: Perform both CGF and MLST analysis on all isolates. For CGF, employ multiplex PCRs targeting the selected accessory genes (e.g., 8 multiplex PCRs each targeting 5 loci for CGF40) [4]. For MLST, follow standard protocols for amplification and sequencing of housekeeping genes.
Data Analysis:
- Calculate Simpson's Index of Diversity (ID) for both methods to compare discriminatory power [4].
- Compute Wallace coefficients to assess concordance between typing methods [4].
- Analyze the ability of each method to differentiate within prevalent sequence types (e.g., ST21 and ST45 for C. jejuni) [4].
Reproducibility Assessment: Repeat CGF analysis on a subset of isolates (e.g., 24) on separate occasions to determine reproducibility, with acceptable concordance thresholds (e.g., >98% identical presence/absence patterns) [5].
The resilience of bacterial subtyping methods to challenging sample conditions reveals a complementary relationship between CGF and MLST. MLST, particularly when implemented using mapping-based approaches from WGS data, demonstrates superior resilience to mixed samples and low sequencing coverage, successfully deriving types in approximately 90% of problematic samples [43]. Conversely, CGF provides higher discriminatory power (ID 0.994 vs. 0.935 for MLST) [4], potentially offering better strain differentiation during outbreak investigations. Method selection should therefore be guided by primary application requirements: MLST mapping-based approaches for suboptimal samples and population studies, and CGF for high-resolution differentiation of closely related strains in well-characterized samples. An integrated strategy, leveraging the respective strengths of each method, provides the most robust framework for comprehensive bacterial subtyping in public health and research contexts.
Evaluating Computational Performance and Resource Requirements
Molecular subtyping of bacterial pathogens is a cornerstone of public health epidemiology, enabling outbreak detection, source tracking, and surveillance of foodborne illnesses. For Campylobacter jejuni and C. coli – leading causes of bacterial gastroenteritis worldwide – the choice of subtyping method significantly impacts the effectiveness and efficiency of epidemiological investigations [4] [28]. Two prominent methods have emerged for characterizing these pathogens: Multilocus Sequence Typing (MLST), which sequences approximately 450-500 base pair fragments of seven housekeeping genes, and Comparative Genomic Fingerprinting (CGF), which detects the presence or absence of 40 accessory genes distributed across the genome using multiplex PCR [4] [6].
This guide provides a systematic comparison of the computational performance, resource requirements, and practical implementation of CGF and MLST within bacterial subtyping research. The evaluation encompasses laboratory workflows, analytical pipelines, discriminatory power, and infrastructure demands to inform method selection for public health laboratories and research institutions engaged in enteric pathogen surveillance.
Experimental Protocols and Methodologies
Comparative Genomic Fingerprinting (CGF) Workflow
The CGF40 method employs an eight-plex PCR approach to detect 40 target genes identified through comparative genomic analyses [4]. The experimental protocol begins with genomic DNA extraction using commercial purification kits. For the CGF40 assay, primers are designed to target regions free of single-nucleotide polymorphisms to ensure specific amplification across diverse C. jejuni strains. The 40 target genes are amplified across eight multiplex PCR reactions, each targeting five loci. Amplification products are subsequently separated by capillary electrophoresis and analyzed for presence/absence patterns to generate a binary fingerprint for each isolate [4]. The CGF type is determined based on the specific combination of detected genes, which can be compared against a database of known profiles.
Multilocus Sequence Typing (MLST) Workflow
The standard MLST protocol for C. jejuni involves amplification and sequencing of approximately 450-500 base pair internal fragments of seven housekeeping genes (aspA, glnA, gltA, glyA, pgm, tkt, and uncA) [4] [6]. Following genomic DNA extraction, each gene fragment is individually amplified via PCR using validated primer sets. Amplification products are purified and sequenced using Sanger sequencing technology with the same primers used for amplification. The resulting sequences are compared to existing allele libraries in the Campylobacter MLST database (http://pubmlst.org/campylobacter/), with unique sequences assigned new allele numbers. The combination of alleles across the seven loci defines the sequence type (ST), which can be further grouped into clonal complexes based on shared ancestry [6].
Validation Framework Using Whole Genome Sequencing
Recent studies have established whole genome sequence (WGS) data as a gold standard for evaluating molecular typing methods [6]. This validation framework involves sequencing a diverse collection of C. jejuni and C. coli isolates using next-generation sequencing platforms. The resulting WGS data undergoes quality assessment through measures like read depth and genome coverage. A reference phylogeny is inferred from 389 highly conserved core (HCC) genes using maximum likelihood methods. In silico derivations of MLST types, CGF types, and other molecular types are compared against this reference phylogeny using statistical measures like the adjusted Wallace coefficient to assess concordance with true strain relationships [6].
Performance Metrics and Comparative Analysis
Discriminatory Power and Typing Resolution
Multiple studies have demonstrated CGF's superior discriminatory power compared to MLST. In a comprehensive analysis of 412 C. jejuni isolates from agricultural, environmental, retail, and human clinical sources, CGF40 exhibited significantly higher resolution with a Simpson's index of diversity of 0.994 compared to 0.935 for MLST sequence types and 0.873 for MLST clonal complexes [4]. This enhanced discrimination is particularly valuable for distinguishing within prevalent sequence types such as ST21 and ST45, where MLST may group epidemiologically unrelated isolates [4].
Table 1: Comparative Analysis of Discriminatory Power Between CGF and MLST
| Method | Number of Loci | Simpson's Index of Diversity | Primary Genetic Target | Typing Output |
|---|---|---|---|---|
| CGF40 | 40 gene presence/absence targets | 0.994 [4] | Accessory genome | Binary fingerprint pattern |
| MLST (ST) | 7 housekeeping genes | 0.935 [4] | Core genome | Sequence type (ST) |
| MLST (CC) | 7 housekeeping genes | 0.873 [4] | Core genome | Clonal complex (CC) |
Computational and Resource Requirements
The implementation of CGF and MLST necessitates distinct laboratory infrastructures, technical expertise, and computational resources. CGF utilizes multiplex PCR and capillary electrophoresis, technologies readily available in molecular biology laboratories, with lower per-sample consumable costs compared to sequencing-based methods. In contrast, MLST requires Sanger sequencing capabilities and access to sequence analysis software and databases, representing higher operational costs but benefiting from extensive standardization and global data sharing through curated databases [4] [28] [6].
Table 2: Resource Requirements and Technical Considerations for CGF and MLST
| Parameter | CGF | MLST |
|---|---|---|
| Equipment Needs | Thermal cycler, capillary electrophoresis system | Thermal cycler, Sanger sequencing platform |
| Technical Expertise | Molecular biology, PCR optimization | Molecular biology, sequencing, bioinformatics |
| Time to Result | ~1-2 days | ~3-5 days |
| Data Portability | Binary profiles (easily shared) | Nucleotide sequences (standardized sharing via pubmlst.org) |
| Cost per Sample | Lower (primarily PCR reagents) | Higher (sequencing reagents, purification) |
| Primary Analysis Software | Fragment analysis software | Sequence assembly, BLAST, MLST database |
| Method Flexibility | Fixed gene target panel | Adaptable to different species with modified schemes |
Concordance with Genomic Phylogeny
When assessed against whole genome sequence-based phylogenies, both CGF and MLST demonstrate strong concordance with true strain relationships, outperforming single-locus methods like flaA SVR typing and porA sequencing [6]. The adjusted Wallace coefficient analyses reveal that MLST and CGF provide complementary insights into strain relationships, with MLST capturing evolutionary relationships through conserved housekeeping genes and CGF reflecting genomic diversity through accessory gene content [6]. This concordance supports the use of both methods for different epidemiological applications, with CGF potentially offering advantages in outbreak settings requiring high resolution.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents and Materials for CGF and MLST Implementation
| Reagent/Material | Function | Application |
|---|---|---|
| PureGene Genomic DNA Purification Kit | Extraction of high-quality genomic DNA from bacterial cultures | Both CGF and MLST [4] [28] |
| CGF40 Primer Sets | Eight multiplex PCR reactions targeting 40 accessory genes | CGF-specific [4] |
| MLST Primer Sets | Amplification of seven housekeeping gene fragments | MLST-specific [4] [6] |
| Montage PCR Centrifugal Filter Devices | Purification of amplification products prior to sequencing | Primarily MLST [4] [28] |
| BigDye Terminator Sequencing Chemistry | Sanger sequencing of amplified gene fragments | MLST-specific [4] |
| Capillary Electrophoresis System | Separation and detection of amplified fragments | CGF-specific [4] |
| ABI Genetic Analyzer | Fragment analysis or sequencing detection | Both CGF and MLST [4] |
The comparative analysis of CGF and MLST reveals distinct advantages and limitations for each method in bacterial subtyping research. CGF offers superior discriminatory power, faster turnaround times, and lower operational costs, making it particularly suitable for outbreak investigations and surveillance activities requiring high resolution between closely related strains [4] [28]. MLST provides robust evolutionary context, extensive standardized databases, and better concordance with core genome phylogeny, making it valuable for long-term epidemiological studies and population genetics analyses [6].
The choice between these methods ultimately depends on the specific research objectives, available resources, and epidemiological context. For public health laboratories with limited sequencing capabilities but requiring rapid, high-resolution subtyping, CGF represents an optimal solution. For research institutions engaged in global surveillance and evolutionary studies, MLST offers unparalleled data portability and phylogenetic context. As whole genome sequencing becomes increasingly accessible, both methods will continue to play important roles in validating and interpreting genomic data for public health action.
This guide provides an objective comparison of two primary bacterial subtyping methods—Comparative Genomic Fingerprinting (CGF) and Multilocus Sequence Typing (MLST)—focusing on critical software selection criteria for researchers and drug development professionals.
Core Experimental Protocols
Multilocus Sequence Typing (MLST) is a standardized method for characterizing bacterial isolates. The protocol involves:
- DNA Extraction and Purification: Genomic DNA is prepared from bacterial isolates using commercial kits [4].
- PCR Amplification: Specific primers are used to amplify seven essential housekeeping genes [4].
- Sequencing and Analysis: Purified PCR products are sequenced using technologies like Sanger sequencing. Sequences are assembled, checked for quality, and compared to a central database to assign allele numbers and Sequence Types (STs) [4].
Comparative Genomic Fingerprinting (CGF) is a higher-resolution, genomics-based method. A validated 40-gene assay (CGF40) involves:
- Marker Selection: Typing markers are selected from accessory genomic regions based on criteria including genomic distribution, variability across strains, and presence/absence patterns identified via genomic surveys [4].
- Multiplex PCR Assay Design: SNP-free PCR primers are designed for each target and assembled into multiplex PCRs (e.g., 8 multiplexes for 5 loci each) [4].
- Fingerprint Analysis: The presence or absence of the target genes is detected to generate a strain-specific fingerprint, without the need for sequencing [4].
Performance Comparison Data
The table below summarizes a direct comparative validation study of CGF40 versus MLST using 412 C. jejuni isolates from various sources [4].
| Feature | Multilocus Sequence Typing (MLST) | Comparative Genomic Fingerprinting (CGF40) |
|---|---|---|
| Typing Basis | Nucleotide sequences of 7 housekeeping genes [4] | Presence/absence of 40 accessory genomic genes [4] |
| Primary Data Output | Allele profiles and Sequence Types (STs) [4] | Binary fingerprint patterns [4] |
| Discriminatory Power (Simpson's Index) | ST Level: 0.935Clonal Complex Level: 0.873 [4] | 0.994 [4] |
| Best Application Context | Long-term epidemiological and population structure studies [4] | Short-term outbreak investigations and surveillance requiring high resolution [4] |
| Key Advantage | High portability, standardized scheme, global databases [4] | Superior ability to differentiate closely related isolates within common STs [4] |
Software Implementation and Data Analysis
Installation and Dependency Management
For researchers, the choice between implementing a bioinformatics pipeline locally or using a web-based platform significantly impacts installation and dependency management.
Local Pipeline Implementation:
- This typically requires working with command-line tools, managing multiple programming languages, and maintaining computing resources, which can be a major bottleneck [48].
- Dependency management involves ensuring all software tools, libraries, and databases are correctly installed and updated.
Web-Based Platforms (e.g., Galaxy @Sciensano):
- Platforms like Galaxy @Sciensano offer a user-friendly solution by providing a curated set of bioinformatics tools through a web browser, eliminating complex local installations [48].
- This instance offers over 50 custom pipelines for complete bacterial isolate characterization, including quality control, assembly, typing, and AMR prediction [48].
- Dependency and Database Management: A key feature is the automated weekly synchronization of 66 databases (e.g., PubMLST, ResFinder). This ensures that analyses use the most current information on sequence types and resistance genes without manual intervention [48].
Advanced Typing and Data Integration
Strain-level resolution is increasingly critical in clinical diagnostics. Metagenomic Next-Generation Sequencing (mNGS) allows for culture-independent subtyping. Tools like the Metagenomic Intra-Species Typing (MIST) software reduce the required sequencing depth by integrating strain-specific Single Nucleotide Polymorphisms (SNPs) and gene content information, enabling strain-level delineation from complex samples like bronchoalveolar lavage fluid [49].
The data output from these methods must be traceable and interpretable. Reputable pipelines address this by generating interactive HTML reports that include key findings, tool parameters, version numbers, and database update dates, which is essential for work under ISO accreditation [48].
Research Reagent Solutions
The table below details key reagents, tools, and databases essential for implementing bacterial subtyping workflows.
| Research Reagent / Solution | Function in Subtyping Workflow |
|---|---|
| PureGene DNA Purification Kit | Genomic DNA preparation from bacterial isolates for MLST sequencing and other PCR-based methods [4]. |
| Illumina NextSeq Platform | High-throughput sequencing for whole-genome sequencing (WGS) and metagenomic NGS (mNGS) applications [49]. |
| PubMLST / EnteroBase Databases | International databases for assigning allele numbers and Sequence Types (STs) in MLST analysis [48]. |
| Comprehensive Antibiotic Resistance Database (CARD) | Reference database for annotating and predicting antimicrobial resistance (AMR) genes from genomic data [49]. |
| ResFinder Database | Database specifically focused on genes and mutations associated with antimicrobial resistance, often synchronized automatically in analysis platforms [48]. |
| MIST Software | Bioinformatic tool for strain-level typing from mNGS data by combining SNP and gene content signals [49]. |
| Galaxy Platform | Web-based, user-friendly interface that provides access to a wide range of curated bioinformatics tools and pipelines, simplifying data analysis [48]. |
Workflow Comparison: CGF vs. MLST
The following diagram illustrates the core procedural steps and data flow for both CGF and MLST methods, highlighting their key differences.
In the field of molecular epidemiology, bacterial strain typing is fundamental for tracking outbreaks, identifying sources of infection, and understanding transmission dynamics. However, researchers frequently encounter a significant challenge: discordant results when applying different typing methods to the same set of bacterial isolates. Such discrepancies can obscure epidemiological relationships and complicate public health responses. This guide focuses on resolving conflicts between two prominent typing methods: Comparative Genomic Fingerprinting (CGF) and Multilocus Sequence Typing (MLST). While both methods provide valuable insights, they target different aspects of bacterial genomes and can produce conflicting results regarding strain relatedness [28] [38]. Understanding the sources of these discrepancies and establishing frameworks for their resolution is crucial for accurate epidemiological interpretation. This article objectively compares the performance of CGF and MLST, provides experimental data supporting these comparisons, and offers practical guidance for researchers navigating conflicting typing results.
Understanding the Fundamental Differences Between CGF and MLST
CGF and MLST operate on distinct principles, targeting different genomic elements and providing complementary yet potentially conflicting information about bacterial relationships.
Multilocus Sequence Typing (MLST) is a sequence-based method that characterizes bacterial strains based on the sequences of approximately 450-500 base pair internal fragments of seven housekeeping genes [50] [29]. These genes are selected for their stability and essential metabolic functions. Strains with identical sequences across all seven loci are assigned the same Sequence Type (ST), which can be further grouped into clonal complexes based on shared alleles [29]. MLST provides excellent reproducibility and portability through centralized databases like PubMLST but has limited discriminatory power due to its focus on a small, conserved portion of the genome [50] [11].
Comparative Genomic Fingerprinting (CGF), in contrast, leverages the presence or absence of accessory genomic elements to generate strain-specific fingerprints [38] [5]. Typically targeting 40-83 accessory genes, CGF captures variability in the more flexible components of the genome [38] [5]. This method utilizes multiplex PCR to detect these variable genes, producing binary profiles that offer high resolution between closely related strains [38]. CGF was specifically designed to offer higher discriminatory power than MLST while maintaining throughput for routine surveillance [28] [38].
The core distinction lies in their genomic targets: MLST indexes variation in essential, slow-evolving housekeeping genes, while CGF probes the accessory genome, which evolves more rapidly through gene gain, loss, and horizontal transfer [28] [5]. This fundamental difference explains why these methods can yield discordant results when assessing the same bacterial isolates.
Quantitative Performance Comparison: CGF vs. MLST
Direct comparative studies provide empirical data on the performance characteristics of CGF and MLST, highlighting their respective strengths and limitations for different epidemiological applications.
Table 1: Performance Metrics of CGF versus MLST for Bacterial Subtyping
| Performance Characteristic | CGF (CGF40 assay) | MLST |
|---|---|---|
| Discriminatory Power (Simpson's Index) | 0.994 [38] | 0.935 (ST level) [38] |
| Typing Resolution | High resolution within prevalent STs [38] | Lower resolution within clonal complexes [28] |
| Technical Basis | Presence/absence of 40 accessory genes [38] [5] | Sequences of 7 housekeeping genes [29] |
| Epidemiological Concordance | High for outbreak detection [28] | Useful for phylogenetic studies [29] |
| Throughput | High, suitable for large-scale surveillance [5] | Lower, more resource-intensive [5] |
| Cost | Lower [5] | Higher [50] |
A study on Campylobacter jejuni directly compared these methods, demonstrating CGF's superior ability to differentiate strains that appeared identical by MLST. The CGF40 assay achieved a Simpson's Index of Diversity of 0.994 compared to 0.935 for MLST at the sequence type level [38]. This enhanced discrimination is particularly valuable for distinguishing strains within prevalent sequence types like ST21 and ST45, which MLST groups together despite potential epidemiological differences [38].
Another critical finding comes from the observation that isolates with identical MLST profiles often comprise isolates with distinct but highly similar CGF profiles [38]. This phenomenon explains a common source of discordance: CGF may differentiate strains that MLST deems identical, providing finer resolution for local outbreak investigations. Conversely, the high concordance between methods, as evidenced by high Wallace coefficients, confirms that both methods generally identify the same broad population structure, albeit at different resolutions [38].
Experimental Protocols for Method Comparison
To ensure valid comparisons between CGF and MLST, researchers should follow standardized protocols for both methods when conducting comparative studies.
CGF (CGF40) Methodology
The CGF40 assay for C. jejuni employs a single multiplex PCR targeting 40 accessory genes, followed by capillary electrophoresis to detect amplified products [38]. The experimental workflow proceeds as follows:
- DNA Extraction: Use a standardized genomic DNA purification kit (e.g., PureGene) for all isolates to ensure consistent quality and concentration [28].
- Multiplex PCR: Perform a single multiplex PCR reaction containing primers for all 40 target accessory genes. These genes are selected from comparative genomic analyses to represent variably present genomic elements [38] [5].
- Amplification Detection: Separate PCR products by capillary electrophoresis to generate binary presence/absence profiles for each target gene.
- Profile Analysis: Analyze electrophoregram data using specialized software to assign CGF types based on the combined pattern of gene presence/absence across all 40 loci [38].
The entire CGF process is designed for high throughput, with lower per-isolate cost and faster turnaround time compared to MLST, making it suitable for analyzing large isolate collections during outbreak investigations [5].
MLST Methodology
The standard MLST protocol for C. jejuni involves sequencing internal fragments of seven housekeeping genes according to established protocols [28]:
- DNA Extraction: Use the same standardized DNA extraction method as for CGF to maintain consistency [28].
- PCR Amplification: Perform individual PCR reactions for each of the seven housekeeping genes (aspA, glnA, gltA, glyA, pgm, tkt, uncA) using primers and conditions archived on the Campylobacter MLST website (http://pubmlst.org/campylobacter/) [28].
- DNA Sequencing: Clean PCR products and sequence both strands using Sanger sequencing with the same primers used for amplification [28].
- Sequence Type Assignment: Compare sequences to those in the MLST database to assign allele numbers and determine the sequence type (ST) based on the combination of all seven alleles [28].
The critical methodological difference lies in CGF's focus on accessory gene presence/absence versus MLST's analysis of sequence variations in core genes. This fundamental distinction in genomic targets drives the potential for discordant results.
Figure 1: Comparative Workflows of CGF and MLST Methods. The diagram illustrates the parallel processes for both typing methods from initial DNA extraction to final type assignment, highlighting their distinct analytical approaches.
Resolution Framework for Discordant Results
When CGF and MLST produce conflicting results regarding strain relatedness, researchers can apply a systematic framework to resolve these discrepancies and arrive at epidemiologically meaningful conclusions.
Interpret Discordance Through Genomic Biology
Discordant results often reflect real biological phenomena rather than methodological errors:
- Accessory Genome Dynamics: CGF's detection of differences in accessory gene content can reveal recent horizontal gene transfer events that MLST would not capture [28] [5]. Such transfers can occur without altering the core genome backbone tracked by MLST.
- Varying Evolutionary Rates: Housekeeping genes targeted by MLST evolve slowly through point mutations, while the accessory genome evolves more rapidly through gene gain and loss, allowing CGF to detect more recent divergence [28].
- Phylogenetic Depth: MLST excels at identifying long-term evolutionary relationships (clonal complexes), while CGF provides higher resolution for recent transmission events within these complexes [38].
Implement Hierarchical Typing Strategies
Studies recommend a hierarchical approach to resolve typing conflicts:
- Use MLST for Broad Classification: Begin with MLST to establish the broad phylogenetic context and clonal complex assignment [28].
- Apply CGF for Fine-Scale Differentiation: Use CGF to differentiate strains within prevalent sequence types, particularly when investigating suspected outbreaks [28] [38].
- Supplement with Additional Markers: For persistent discordance, consider supplementary methods such as flaA SVR sequencing or porA typing to provide additional resolution [28].
This combined approach leverages the strengths of both methods, with MLST providing phylogenetic context and CGF enabling high-resolution discrimination of recently diverged strains [28].
Integrate Epidemiological Context
Always interpret typing results alongside epidemiological data:
- Temporal Patterns: Strains with identical CGF profiles isolated within short timeframes are more likely to be epidemiologically linked, regardless of MLST results [28].
- Spatial Distribution: Geographical clustering of specific CGF types can reveal transmission pathways that MLST alone might miss [38].
- Source Attribution: CGF has demonstrated utility in distinguishing strains from different reservoirs (e.g., human vs. animal) within the same MLST type [38].
Figure 2: Decision Framework for Resolving Discordant CGF and MLST Results. This workflow provides a systematic approach to interpreting conflicting typing results by integrating methodological strengths with epidemiological context.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation and comparison of CGF and MLST methods require specific laboratory reagents and computational resources. The following table details essential materials for conducting these analyses.
Table 2: Essential Research Reagents and Materials for CGF and MLST Typing
| Item | Function/Application | Specific Examples/Notes |
|---|---|---|
| Genomic DNA Purification Kit | Standardized DNA extraction for both CGF and MLST | PureGene kit (Gentra Systems) or equivalent [28] |
| PCR Reagents | Amplification of target genes | PuReTaq Ready-to-Go PCR beads (Amersham BioSciences) or commercial PCR kits (Applied Biosystems) [28] [51] |
| Capillary Electrophoresis System | Separation and detection of CGF PCR products | Required for CGF analysis to generate binary profiles [38] |
| DNA Sequencer | Sanger sequencing for MLST | ABI PRISM systems with BigDye terminator kits [28] [51] |
| Specialized Software | Data analysis and profile comparison | BioNumerics, GelCompar for pattern analysis; PubMLST for sequence type assignment [28] [11] |
| Reference Databases | Strain comparison and data sharing | PubMLST.org for MLST; CGF databases for profile comparison [28] [29] |
Discordant results between CGF and MLST typing methods present both challenges and opportunities in bacterial subtyping research. Rather than representing methodological failure, such discordance often reveals important biological phenomena, including differential evolutionary rates between core and accessory genomes and recent horizontal gene transfer events. The resolution framework presented here emphasizes hierarchical interpretation—using MLST for broad phylogenetic classification and CGF for fine-scale outbreak detection—supplemented by robust epidemiological context. As the field continues to evolve toward whole-genome sequencing, understanding how to resolve conflicts between established methods like CGF and MLST remains crucial for accurate epidemiological investigations and effective public health responses.
Head-to-Head Performance: Validating Typing Methods Against Real Outbreak Data
Molecular subtyping is a cornerstone of modern bacterial outbreak investigation, enabling researchers to distinguish between bacterial strains for source tracking and transmission route identification. This guide provides an objective comparison of three prominent subtyping methods—Comparative Genomic Fingerprinting (CGF), Multilocus Sequence Typing (MLST), and Pulsed-Field Gel Electrophoresis (PFGE)—evaluating their discriminatory power, technical requirements, and applicability in outbreak settings. Based on comparative studies across multiple bacterial pathogens, we present experimental data demonstrating that CGF generally provides superior discrimination for outbreak detection, while MLST offers better phylogenetic context, and PFGE serves as an established gold standard with high reproducibility. This analysis aims to equip researchers, scientists, and drug development professionals with evidence-based guidance for selecting appropriate subtyping methods based on their specific investigative needs and technical capabilities.
Bacterial subtyping methods are essential tools for detecting outbreaks, investigating transmission dynamics, and implementing effective public health interventions. The discriminatory power of a subtyping method—its ability to differentiate between epidemiologically unrelated strains—varies significantly across techniques and directly impacts outbreak detection sensitivity [50]. PFGE has long been considered the gold standard for outbreak investigations due to its high reproducibility and established protocols within networks like PulseNet International [52] [11]. However, sequence-based methods like MLST and CGF are increasingly utilized for their superior portability and phylogenetic capabilities [53].
MLST characterizes bacterial isolates based on nucleotide sequences of internal fragments of typically seven housekeeping genes, assigning allelic profiles to define sequence types (STs) [53]. This method provides excellent reproducibility and phylogenetic relevance but may lack sufficient discrimination for some outbreak investigations [50]. CGF, a more recently developed method, utilizes multiplex PCR to detect presence or absence of genes identified through comparative genomics as having high intraspecies variability, potentially offering enhanced discrimination while maintaining technical feasibility [28].
This guide systematically compares the discriminatory power of CGF, MLST, and PFGE through direct experimental comparisons across multiple bacterial pathogens and outbreak scenarios, providing researchers with practical insights for method selection in different investigative contexts.
Fundamental Principles and Techniques
Pulsed-Field Gel Electrophoresis (PFGE)
PFGE involves digesting bacterial genomic DNA with restriction enzymes that recognize rare cutting sites, generating large DNA fragments (20-800 kb) that are separated using alternating electric fields [52] [11]. The resulting banding patterns serve as DNA fingerprints for strain comparison. Standardized protocols exist for multiple bacterial pathogens, with restriction enzymes like XbaI commonly used for Salmonella and SpeI for Pseudomonas aeruginosa [11] [54]. Pattern analysis typically uses software like BioNumerics or GelCompar, with isolates showing ≥87% similarity often considered genetically related [54].
Multilocus Sequence Typing (MLST)
MLST sequences approximately 450-500 bp internal fragments of typically seven housekeeping genes to identify allelic variations [53]. Each unique allele receives a number, and the combination of alleles across loci defines the sequence type (ST). The method leverages online databases like PubMLST for global comparison and curation of allelic profiles [53]. MLST data are highly portable between laboratories and provide stable references for long-term epidemiological monitoring and phylogenetic studies [55] [53].
Comparative Genomic Fingerprinting (CGF)
CGF utilizes multiplex PCR to amplify multiple genetic loci identified through comparative genomic analyses as having high intraspecies variability [28]. Unlike MLST, which focuses on conserved housekeeping genes, CGF targets genomic regions with natural variation that can provide enhanced discrimination. The presence or absence patterns of these amplifications create fingerprints for strain differentiation. CGF methods have been developed for several pathogens including Campylobacter jejuni and C. coli [28].
Visual Comparison of Fundamental Methodologies
The diagram below illustrates the core procedural differences between PFGE, MLST, and CGF workflows:
Comparative Performance Analysis
Direct Comparison Studies
Campylobacter jejuni and C. coli Analysis
A comprehensive sentinel site surveillance study compared five subtyping methods for Campylobacter outbreak detection [28]. The study evaluated PFGE, MLST, flaA SVR sequencing, porA sequencing, and CGF using 440 human and non-human isolates. Researchers found that CGF demonstrated optimal performance for detecting epidemiologically relevant clusters, while MLST combined with flaA SVR sequencing provided complementary value for identifying populations linked to specific infection sources [28].
Table 1: Method Performance in Campylobacter Surveillance Study
| Method | Discriminatory Ability | Epidemiologic Concordance | Best Application |
|---|---|---|---|
| CGF | Highest | High | Primary outbreak detection |
| PFGE | High | High | Outbreak confirmation |
| MLST + flaA SVR | Moderate | Moderate | Population structure analysis |
| MLST alone | Lower | Lower | Phylogenetic studies |
Pseudomonas aeruginosa Investigation
A comparison of PFGE and MLST for 90 P. aeruginosa isolates from intensive care unit surveillance demonstrated PFGE's superior discriminatory power [54]. Using Simpson's index of diversity (D), PFGE (D=0.999) outperformed MLST (D=0.975), identifying 85 distinct types compared to 60 sequence types. However, MLST provided better detection of genetic relatedness through clonal complex analysis, highlighting the method's value for understanding long-term transmission patterns [54].
Listeria monocytogenes Evaluation
A study of 175 L. monocytogenes strains compared serotyping, PFGE, and MLST based on six gene loci (actA, betL, hlyA, gyrB, pgm, recA) [55]. MLST identified 122 sequence types, demonstrating better differentiation of most strains compared to PFGE. The discriminating ability of PFGE exceeded serotyping but was inferior to MLST for certain strains, particularly those with identical PFGE patterns but different origins [55].
Quantitative Discrimination Metrics
Table 2: Comparative Method Performance Across Multiple Pathogens
| Pathogen | PFGE Discrimination | MLST Discrimination | CGF Discrimination | Reference |
|---|---|---|---|---|
| Pseudomonas aeruginosa | 0.999 (Simpson's Index) | 0.975 (Simpson's Index) | Not tested | [54] |
| Campylobacter spp. | High (comparable to CGF) | Moderate | Highest for outbreak detection | [28] |
| Listeria monocytogenes | Lower than MLST for some strains | Higher than PFGE (122 STs) | Not tested | [55] |
| Salmonella Enteritidis | Lower (45% share single pattern) | Moderate | Higher than PFGE (inferred) | [56] |
Technical Considerations and Method Selection
CGF Implementation and Workflow
The CGF method for Campylobacter utilizes a multiplex PCR approach targeting genomic loci identified through comparative genomic hybridization studies [28]. The experimental protocol involves:
- DNA extraction using commercial kits (e.g., PureGene genomic DNA purification kit)
- Multiplex PCR amplification with primers designed to target highly variable genomic regions
- Fragment analysis using capillary electrophoresis to separate amplification products
- Pattern analysis and classification using specialized software to generate CGF types [28]
This method provides technical accessibility for laboratories already equipped for conventional PCR and fragment analysis, without requiring massive sequencing infrastructure.
Integrated Selection Framework
Method choice depends on investigation objectives:
- Outbreak detection and cluster identification: CGF or PFGE provide optimal discrimination
- Long-term phylogenetic studies and population genetics: MLST offers superior stability and portability
- Routine surveillance with established infrastructure: PFGE maintains advantages through standardized protocols and extensive databases
- Comprehensive outbreak investigation: Combining methods (e.g., MLST for broad classification followed by CGF or PFGE for high-resolution discrimination) often provides the most complete epidemiological picture [28]
Research Toolkit for Bacterial Subtyping
Essential Research Reagents and Equipment
Table 3: Essential Research Reagents and Equipment for Subtyping Methods
| Category | Specific Items | Application | Method |
|---|---|---|---|
| DNA Extraction | PureGene genomic DNA purification kit, Prepman Ultra | Nucleic acid isolation | All methods |
| Restriction Enzymes | XbaI, SpeI, NotI, SfiI | DNA digestion for fingerprinting | PFGE |
| Electrophoresis | CHEF DR apparatus, agarose gels, molecular size standards | DNA separation | PFGE |
| PCR Reagents | Primers, DNA polymerase, dNTPs, buffer systems | Target amplification | MLST, CGF |
| Sequencing | BigDye terminators, capillary sequencers | Nucleotide determination | MLST |
| Fragment Analysis | Capillary electrophoresis systems, size standards | Amplification product separation | CGF |
| Analysis Software | BioNumerics, GelCompar, Fingerprinting II | Pattern analysis and comparison | All methods |
Standardized PFGE Protocol
Based on PulseNet International standards for Listeria monocytogenes [55]:
- DNA preparation: Embed bacterial cells in agarose plugs and treat with proteinase K
- Restriction digestion: Digest DNA with AscI restriction enzyme at 37°C for 4 hours
- Electrophoresis: Separate fragments using CHEF DR II apparatus with pulse times from 4.0-40.01 seconds over 16 hours at 180V
- Pattern analysis: Compare banding patterns to reference standards and database patterns
MLST Protocol forL. monocytogenes
As implemented by [55] for six gene loci:
- Primer design: Design primers in conserved regions flanking variable segments of target genes (actA, betL, hlyA, gyrB, pgm, recA)
- PCR amplification: Amplify target fragments using standardized conditions
- DNA sequencing: Sequence both strands using amplification primers
- Allele assignment: Compare sequences to existing alleles in database, assign allele numbers and sequence types
The CGF workflow involves multiple parallel processes that converge to generate high-resolution strain fingerprints:
The comparative analysis of CGF, MLST, and PFGE reveals a complex landscape where each method offers distinct advantages for bacterial subtyping in outbreak settings. CGF emerges as particularly valuable for outbreak detection and investigation of common pathogens like Campylobacter, where its design targeting variable genomic regions provides optimal discrimination of closely related strains [28]. MLST provides unparalleled stability and phylogenetic context, making it ideal for long-term epidemiological studies and global surveillance networks [53]. PFGE maintains utility as a proven gold standard with extensive databases and standardized protocols, though its transition toward replacement by whole-genome sequencing-based methods is underway [52] [11].
The choice of subtyping method should be guided by specific investigation needs: CGF for high-resolution outbreak detection where available, MLST for phylogenetic and population studies, and PFGE for routine surveillance within established networks. As sequencing technologies continue to advance and become more accessible, whole-genome sequencing is poised to potentially supplant all these methods for comprehensive outbreak investigation [56] [50]. However, for laboratories without immediate access to whole-genome sequencing capabilities, CGF, MLST, and PFGE remain powerful, validated approaches for bacterial subtyping in outbreak settings.
Each method contributes uniquely to our understanding of bacterial transmission dynamics, and their complementary use often provides the most comprehensive insight into outbreak sources and spread. Researchers should consider their specific technical capabilities, investigative timelines, and discrimination requirements when selecting the most appropriate subtyping approach for their particular context.
Molecular subtyping of bacterial pathogens is a cornerstone of modern epidemiology, enabling researchers to link cases during outbreaks, identify transmission sources, and understand microbial population dynamics. The central challenge lies in achieving strong epidemiological concordance—where genetic clustering of bacterial strains accurately reflects the clinical and epidemiological links between patient cases. Among the various typing methods available, Multi-Locus Sequence Typing (MLST) and Comparative Genomic Fingerprinting (CGF) have emerged as prominent techniques, each with distinct advantages and limitations. MLST, based on sequence analysis of a small set of housekeeping genes, provides a standardized, portable approach for phylogenetic studies and long-term surveillance. In contrast, CGF, which targets presence or absence patterns in numerous accessory genes, offers higher resolution for distinguishing closely related strains, potentially offering superior alignment with outbreak epidemiology. This guide objectively compares the performance of CGF and MLST, focusing on their ability to generate bacterial clusters that concord with patient data, thereby aiding researchers and drug development professionals in selecting the optimal subtyping method for their specific public health and research objectives.
Methodological Foundations: CGF vs. MLST
The fundamental differences between CGF and MLST lie in their genomic targets and underlying methodologies. Understanding these technical distinctions is prerequisite for interpreting their performance in epidemiological investigations.
Multi-Locus Sequence Typing (MLST)
MLST is a sequence-based approach that characterizes bacterial strains by indexing the nucleotide sequences of approximately seven housekeeping genes [50]. These genes are essential for basic cellular functions and are typically conserved within a bacterial species. The process involves:
- PCR Amplification: Amplifying the seven defined housekeeping loci.
- DNA Sequencing: Determining the DNA sequences of these amplified fragments.
- Allele and ST Assignment: Comparing the sequences to a centralized database (e.g., PubMLST) to assign an allele number for each locus. The combination of the seven allele numbers defines the Sequence Type (ST) for the isolate [5].
This method is highly reproducible and portable between laboratories, making it excellent for global surveillance and phylogenetic studies of population structure. However, because it targets a small number of stable core genes, its discriminatory power is limited [50].
Comparative Genomic Fingerprinting (CGF)
CGF is a gene presence/absence-based method that leverages variability in the accessory genome—genes not shared by all strains of a species. The development and application of CGF involve:
- Comparative Genomics: Initially, whole genome sequences of multiple strains are compared to identify a large set of accessory genes [35] [5].
- Marker Selection: Computational tools (e.g., CGF Optimizer) are used to select an optimal subset of genes (e.g., 40 loci for CGF40) that provide high resolution and concordance with a reference phylogeny [5].
- High-Throughput Profiling: A multiplex PCR or similar high-throughput assay is developed to detect the presence or absence of these selected accessory genes in test isolates, generating a binary fingerprint for each strain [35] [5].
CGF targets regions of the genome that are more variable than housekeeping genes, offering the potential for greater discriminatory power to distinguish between closely related isolates, which is often crucial in outbreak settings.
Table 1: Core Methodological Characteristics of MLST and CGF
| Feature | MLST | CGF |
|---|---|---|
| Genomic Target | Core genome (housekeeping genes) | Accessory genome |
| Basis of Discrimination | Nucleotide sequence polymorphisms | Presence/absence of genes |
| Typical Number of Loci | 7 | 40-83 (assay-dependent) |
| Data Output | Sequence Type (ST) | Binary fingerprint |
| Primary Analysis | Sequence alignment and allele calling | Presence/absence scoring |
Workflow Visualization
The following diagram illustrates the key procedural steps for both MLST and CGF methods, highlighting their parallel yet distinct pathways from bacterial isolate to final cluster analysis.
Direct Performance Comparison: Experimental Data
The theoretical advantages of CGF are borne out in direct comparative studies. Research evaluating subtyping methods against a "gold standard" phylogeny built from hundreds of highly conserved core genes provides a rigorous measure of their ability to reflect true strain relationships.
Concordance with a Reference Phylogeny
A seminal study analyzed 104 whole genome sequences of C. jejuni and C. coli to evaluate MLST, flaA typing, porA typing, and CGF. The adjusted Wallace coefficient (AWC) was used to measure concordance with the reference phylogeny, where a value of 1 indicates perfect agreement [35].
The key finding was that both MLST and CGF provided better estimates of the true phylogeny than single-locus methods. This is because both are multi-locus approaches, making them more robust against the confounding effects of horizontal gene transfer and recombination that frequently occur in bacterial genomes [35].
Furthermore, the same framework demonstrated that a CGF40 assay for Arcobacter butzleri achieved an AWC of 1.0 compared to a reference phylogeny based on 72 accessory genes, indicating that the optimized 40-gene assay perfectly recapitulated the clustering of the larger, more comprehensive gene set [5].
Discriminatory Power and Epidemiological Resolution
Discriminatory power is a critical metric, defined as the ability of a method to distinguish between unrelated strains. This is often quantified using Simpson's Index of Diversity (ID).
For A. butzleri, the CGF40 assay demonstrated exceptionally high discriminatory power. Analysis of 156 isolates from diverse sources resulted in 121 distinct CGF profiles and a Simpson's Index of Diversity > 0.969 [5]. This high resolution is vital for detecting subtle differences during outbreak investigations, where distinguishing the outbreak strain from closely related, but epidemiologically unrelated, background strains is essential.
Table 2: Quantitative Performance Comparison of MLST and CGF
| Performance Metric | MLST Performance | CGF Performance | Interpretation and Context |
|---|---|---|---|
| Concordance (AWC) | Good | Excellent | CGF showed perfect concordance (AWC=1.0) with a reference phylogeny in A. butzleri [5]. Both multi-locus methods outperformed single-locus typing [35]. |
| Discriminatory Power (ID) | Moderate to High | Very High | CGF40 for A. butzleri achieved an ID > 0.969, identifying 121 unique profiles among 156 isolates [5]. CGF generally offers better differentiation of closely related strains. |
| Epidemiological Linkage | Can miss links due to limited resolution [35]. | Can identify clades of genetically similar isolates [5]. | CGF's higher resolution improves the detection of outbreak clusters that might appear identical by MLST. |
| Throughput & Cost | Resource-intensive, lower throughput [5]. | High-throughput, lower cost per sample [5]. | CGF is better suited for large-scale surveillance and routine screening due to its simpler, faster workflow. |
Essential Research Toolkit for Bacterial Subtyping
Implementing MLST and CGF methodologies requires a specific set of reagents, equipment, and bioinformatic resources. The following table details key solutions essential for research in this field.
Table 3: Research Reagent Solutions for Molecular Subtyping
| Item Name | Function/Application | Method |
|---|---|---|
| PubMLST Database | Centralized repository for allele sequences and MLST schemes; used for assigning Sequence Types. | MLST [13] [57] |
| CGF Optimizer | Bioinformatics tool for selecting an optimal subset of accessory genes to create a high-resolution CGF assay. | CGF [5] |
| ResFinder/PlasmidFinder | Databases used with BLASTn to identify antimicrobial resistance genes and plasmid replicons within WGS data. | Bioinformatics [57] |
| SPAdes/Shovill Assembler | De novo genome assembly tools used to reconstruct bacterial genomes from short-read sequencing data. | WGS [13] [57] |
| ChewBBACA | A bioinformatics tool for performing core genome MLST (cgMLST) and schema validation, often used in assembly-based approaches. | cgMLST [13] |
The Impact of Bioinformatics on Typing Results
The move towards whole-genome sequencing (WGS) as a gold standard highlights that methodological choices extend beyond the wet lab. Bioinformatic variability in processing WGS data can significantly impact subtyping results, affecting epidemiological concordance.
A 2024 study demonstrated that the choice of assembly tool (e.g., SPAdes, Shovill, Unicycler) can introduce variability in the resulting cgMLST profiles, a finding validated across six different bacterial species including Listeria monocytogenes and Salmonella enterica [13]. This variability is not just tool-related but is also influenced by the intrinsic composition of the bacterial genomes themselves, such as repetitive sequences or regions with extreme GC content [13].
This underscores a critical point for epidemiological concordance: standardized bioinformatics pipelines are as crucial as standardized laboratory protocols. Inconsistent data processing can create artificial genetic differences or mask real ones, leading to misinterpretation of strain relationships and a failure to align genetic clusters with patient data. For reproducible and comparable results, laboratories must harmonize their entire workflow, from DNA extraction to final data analysis [13].
The choice between MLST and CGF for achieving epidemiological concordance hinges on the specific objectives of the study. MLST remains a powerful, standardized tool for global phylogenetic analysis and long-term population surveillance, providing a stable nomenclature for comparing strains across time and geography. However, for high-resolution outbreak investigations where distinguishing between closely related strains is paramount, CGF offers superior discriminatory power, higher throughput, and lower cost. The experimental data confirms that CGF demonstrates excellent concordance with reference phylogenies and can effectively identify clades of genetically similar isolates that are epidemiologically linked. As the field progresses, the integration of whole-genome sequencing and standardized bioinformatics pipelines will ultimately provide the highest resolution for aligning genetic clusters with patient data, but CGF stands out as a highly effective and deployable method for current large-scale public health surveillance and outbreak detection.
The advent of whole-genome sequencing (WGS) has revolutionized bacterial subtyping, enabling high-resolution tracking of pathogen transmission and outbreak dynamics. Core genome multilocus sequence typing (cgMLST) and core single nucleotide polymorphism (coreSNP) analysis have emerged as the leading, next-generation methods for genomic epidemiology. This guide provides a comparative analysis of cgMLST and coreSNP, detailing their methodologies, performance characteristics, and applications within the broader context of molecular typing evolution. Supported by experimental data and standardized protocols, this resource aims to inform researchers and public health professionals in selecting appropriate strategies for investigating bacterial outbreaks and surveillance.
Molecular epidemiology relies on robust typing methods to determine genetic relatedness between bacterial isolates, enabling effective surveillance and outbreak investigation. Traditional methods, such as pulsed-field gel electrophoresis (PFGE) and multilocus sequence typing (MLST), have been foundational but are limited by lower discriminatory power and challenges in standardizing results across laboratories [58] [12]. PFGE, while long considered the "gold standard," suffers from limited portability and an inability to distinguish closely related strains in highly clonal populations [8] [59]. Similarly, conventional MLST, which sequences seven housekeeping genes, often lacks the resolution needed for fine-scale outbreak investigations [58] [12].
The introduction of whole-genome sequencing (WGS) has overcome these limitations, providing unprecedented resolution for subtyping. Two primary WGS-based approaches have emerged: core genome multilocus sequence typing (cgMLST) and core single nucleotide polymorphism (coreSNP) analysis. These methods represent a significant advancement over traditional techniques, offering superior discriminatory power for tracking microbial populations and investigating transmission chains [8] [12] [60].
Methodological Principles
Core Genome Multilocus Sequence Typing (cgMLST)
cgMLST extends the concept of conventional MLST to the genome level. It utilizes a defined scheme of hundreds to thousands of core genes—those present in most (typically 95-99%) members of a bacterial species [12] [61]. The process involves:
- Gene Definition: A standardized scheme is developed, comprising core genes that are stable, under selection pressure, and exclude repetitive elements and pseudogenes [12].
- Allele Calling: For each isolate, each gene in the scheme is compared to a curated database of known alleles and assigned an allele number based on its sequence.
- Profile Comparison: The genetic distance between two isolates is expressed as the number of loci with differing allele numbers [34] [61].
A key advantage of cgMLST is its standardization; it allows for reproducible comparisons across laboratories and over time, facilitating global data exchange [12] [60]. For example, a study on Pseudomonas aeruginosa demonstrated a high correlation (R² of 0.92–0.99) between cgMLST and coreSNP distances, confirming their congruence for outbreak investigation [61].
Core Single Nucleotide Polymorphism (coreSNP) Analysis
coreSNP analysis identifies single nucleotide polymorphisms across the core genome, which includes regions conserved across all isolates of a species. The typical workflow is:
- Reference Selection: A high-quality genome, closely related to the isolates under investigation, is selected as a reference.
- Read Mapping: Sequencing reads from each isolate are mapped to the reference genome.
- Variant Calling: Bioinformatics tools identify high-quality SNP positions where the isolate differs from the reference.
- Phylogenetic Inference: The matrix of coreSNP differences is used to construct phylogenetic trees and assess relatedness [8] [62] [63].
coreSNP analysis offers exceptionally high resolution, making it particularly suitable for distinguishing between highly clonal isolates [8] [62]. However, its results can be influenced by the choice of reference genome and the specific parameters of the bioinformatic pipeline, making standardization between laboratories more challenging [62] [61].
Comparative Performance Analysis
Extensive benchmarking studies have evaluated the performance of cgMLST and coreSNP against traditional methods and each other. The following tables summarize key quantitative comparisons based on published experimental data.
Table 1: Comparative Discriminatory Power of Typing Methods Across Pathogens
| Bacterial Species | Typing Method Comparison | Key Finding | Reference |
|---|---|---|---|
| Klebsiella pneumoniae | cgMLST vs. coreSNP vs. PFGE | cgMLST & coreSNP showed higher discriminant power than PFGE; both suitable for transmission analysis. | [8] |
| Acinetobacter baumannii | cgMLST vs. MLST vs. PFGE | Resolution order: cgMLST > MLST (Oxford) > PFGE > MLST (Pasteur). cgMLST was the most comprehensive. | [59] |
| Salmonella enterica | cgMLST vs. wgMLST vs. SNP | cgMLST was congruent with SNP-based analysis and epidemiological data. | [12] [60] |
| Pseudomonas aeruginosa | cgMLST/wgMLST vs. coreSNP | High correlation (R² 0.92–0.99) between cgMLST and core-SNP distances. | [61] |
| Staphylococcus capitis | cgMLST vs. coreSNP | cgMLST and coreSNP provided comparable resolution (217 vs. 242 distinct genotypes). | [64] |
Table 2: Technical and Operational Comparison of cgMLST and coreSNP
| Characteristic | cgMLST | coreSNP |
|---|---|---|
| Analysis Principle | Gene-by-gene allele comparison | Nucleotide-level variant calling |
| Resolution | High, suitable for outbreak strain discrimination | Very high, can distinguish closely related isolates in a outbreak |
| Standardization & Portability | High (relies on standardized, portable schemes) | Lower (dependent on reference genome and pipeline parameters) |
| Reference Dependency | Low (scheme-based, no single reference required) | High (output sensitive to reference genome choice) |
| Handling of Homologous Recombination | Robust (treats recombinant regions as single allele changes) | Requires specific filtering tools to avoid misleading distances |
| Computational Demand | Moderate | Can be computationally demanding |
| Ease of Data Interpretation | Straightforward (allelic difference thresholds) | Requires phylogenetic interpretation |
| Ideal Use Case | Routine surveillance, inter-laboratory comparisons, database building | High-resolution outbreak investigation, phylogenetic studies |
A 2020 study on Klebsiella pneumoniae concluded that both cgMLST and coreSNP are more discriminant than PFGE and suitable for transmission analysis. However, cgMLST appeared inferior to coreSNP in the phylogenetic reconstruction of the K. pneumoniae CG258 group, with cgMLST wrongly clustering certain strains that were properly distinguished by coreSNP [8]. This highlights that while the methods are often concordant, coreSNP can provide superior phylogenetic insight in specific clonal complexes.
A 2025 study comparing commercial cgMLST pipelines (Ridom SeqSphere+, 1928 Diagnostics, and ARESdb) found that while allelic distances could differ significantly, the final clustering conclusions using suggested species-specific thresholds were highly concordant (100% concordance between SeqSphere+ and 1928, 99.5% with ARESdb) [34]. This underscores that cgMLST is a robust and standardized tool for outbreak detection.
Detailed Experimental Protocols
To ensure reproducibility and provide a framework for implementation, this section outlines standard operating procedures for cgMLST and coreSNP analyses, as applied in cited studies.
Protocol 1: cgMLST Analysis Using Ridom SeqSphere+
This protocol is adapted from studies on Staphylococcus capitis [64] and Acinetobacter baumannii [59].
Wet-Lab Procedure:
- Isolate Purification: Purify bacterial cultures by two successive single colony selections on solid agar medium incubated overnight at 37°C.
- DNA Extraction: Extract genomic DNA using a validated kit (e.g., Maxwell 16 Cell DNA Purification Kit). Assess DNA quality and quantity using spectrophotometry or fluorometry.
- Library Preparation & Sequencing: Prepare sequencing libraries (e.g., with Illumina Nextera XT kit) and sequence on an Illumina platform (e.g., NextSeq500) to generate paired-end reads (e.g., 2x150 bp).
Bioinformatic Analysis:
- Quality Control: Assess raw read quality using tools like FastQC.
- Data Assembly: Perform de novo assembly of reads using SPAdes.
- cgMLST Typing:
- Import assembled genomes into Ridom SeqSphere+ software.
- Select the appropriate, species-specific cgMLST scheme (e.g., 1492 genes for S. capitis [64], 2398 genes for K. pneumoniae [8]).
- The software performs an automated, assembly-based allele call for each locus in the scheme.
- Generate a matrix of pairwise allelic differences between isolates.
- Cluster Analysis: Use the allelic difference matrix to create a minimum spanning tree or perform hierarchical clustering. Apply species-specific thresholds to define clusters (e.g., ≤10 allele differences for E. coli, ≤15 for K. pneumoniae) [34].
Protocol 2: coreSNP Analysis Pipeline
This protocol is based on workflows used for Salmonella enterica [62] and Klebsiella pneumoniae [8].
Wet-Lab Procedure: (Identical to Protocol 1 steps 1-3).
Bioinformatic Analysis:
- Quality Control & Trimming: Use Trimmomatic or similar to remove low-quality bases and adapter sequences from raw reads.
- Reference-Based Mapping:
- Select a closed, high-quality reference genome closely related to the isolates under study.
- Map quality-trimmed reads from each isolate to the reference genome using a tool like BWA-MEM or Bowtie 2.
- Variant Calling:
- Process alignment files (e.g., sort, mark duplicates) using SAMtools.
- Call SNPs using a variant caller like SAMtools/bcftools or GATK. Apply quality filters (e.g., minimum read depth, mapping quality, base quality).
- Extract SNPs located in the core genome, excluding repetitive regions, phage DNA, and recombinant sites (filtered using tools like Gubbins or ClonalFrameML).
- Distance Calculation & Phylogenetics:
- Generate a coreSNP alignment from the filtered, high-quality SNPs.
- Calculate a pairwise SNP distance matrix.
- Construct a phylogenetic tree using maximum-likelihood (e.g., RAxML) or neighbor-joining methods.
Successful implementation of cgMLST and coreSNP typing requires a suite of laboratory and computational resources. The following table details key solutions and their functions.
Table 3: Essential Reagents and Resources for High-Resolution Typing
| Category | Item / Solution | Function / Application | Example(s) |
|---|---|---|---|
| Wet-Lab | DNA Extraction Kit | High-quality, high-molecular-weight genomic DNA isolation | Maxwell 16 Cell DNA Purification Kit [8] |
| Library Preparation Kit | Preparation of sequencing-ready libraries from DNA | Illumina Nextera XT [8] | |
| Sequencing Platform | High-throughput generation of short-read genomic data | Illumina NextSeq500, HiSeq 2000 [8] [59] | |
| Bioinformatic Software | cgMLST Analysis | Standardized allele calling and cluster analysis | Ridom SeqSphere+ [34] [59] [64], BioNumerics [61], 1928 Platform [34] |
| coreSNP Analysis | Read mapping, variant calling, and phylogenetic inference | CFSAN-based workflow [62], PHEnix [62], CSI Phylogeny [62] | |
| Genome Assembler | De novo reconstruction of genomes from sequencing reads | SPAdes [8] [61] | |
| Recombination Filtering | Identification and masking of recombinant genomic regions | Gubbins [63], ClonalFrameML [63] | |
| Databases & Schemes | cgMLST Scheme | Species-specific set of core genes for standardized typing | PubMLST, cgMLST.org [34] |
| Genomic Database | Repository for raw sequencing data and assemblies | NCBI SRA, GenBank [8] [61] |
cgMLST and coreSNP analyses represent a paradigm shift in bacterial subtyping, offering robust, high-resolution alternatives to traditional methods like PFGE and MLST. The choice between them depends on the specific application: cgMLST excels in standardized surveillance and inter-laboratory comparisons due to its portability and ease of data interpretation, while coreSNP provides ultimate resolution for fine-scale phylogenetic investigations of outbreaks involving highly clonal strains. As WGS becomes more accessible, these methods will form the cornerstone of public health microbiology, enabling precise tracking and control of infectious disease threats.
The field of bacterial subtyping for outbreak surveillance and evolutionary research has undergone a significant transformation, moving from traditional, lower-resolution methods to whole-genome sequencing (WGS)-based analysis. Within this context, a critical evaluation of the bioinformatics software used to analyze this genomic data is essential. This guide provides an objective comparison of core genome multi-locus sequence typing (cgMLST) software pipelines and a novel decentralized method against traditional typing results, framing the analysis within a broader thesis on the comparative performance of cgMLST versus traditional multilocus sequence typing (MLST) for bacterial subtyping research. The benchmarking data, experimental protocols, and performance metrics summarized here are intended to assist researchers, scientists, and drug development professionals in selecting appropriate methodologies for their specific applications.
Performance Benchmarking of cgMLST Pipelines and CDST
Comparative Performance of Commercial cgMLST Pipelines
Microbial whole-genome sequencing (WGS)-based methods have largely replaced conventional techniques for genomic relatedness analysis in outbreak investigation and surveillance. Among WGS methods, core genome multi-locus sequence typing (cgMLST) provides a standardized approach for strain comparisons at high resolution. A 2025 study compared three commercial cgMLST software pipelines—Ridom SeqSphere+, 1928 Diagnostics' platform, and Ares Genetics ARESdb—for identifying related strains among 255 isolates of common bacterial pathogens [34].
The study evaluated eight common healthcare-associated infection pathogens: Acinetobacter baumannii, Escherichia coli, Enterococcus faemannii, Enterococcus faecium, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, and Serratia marcescens. Isolates were previously identified as clustered with at least one other isolate from the same patient or different patients. The analysis generated pairwise distance matrices for 6,077 isolate pairs across all three platforms [34].
Table 1: Concordance of Commercial cgMLST Pipelines Using Suggested Clustering Thresholds
| Pipeline | Overall Concordance with SeqSphere+ | Same-Patient Clustered Pairs | Different-Patient Clustered Pairs | Different-Patient Non-Clustered Pairs |
|---|---|---|---|---|
| 1928 Platform | 100% | 100% | 100% | 100% |
| ARESdb | 99.5% | 91.8% | 96.1% | 100% |
Despite high concordance using suggested clustering thresholds, the study found statistically significant differences in mean allelic distances among clustered isolate pairs between the pipelines. ARESdb showed substantially greater allelic distances (mean 7.6±7.17 for same-patient clustered pairs) compared to SeqSphere+ (1.18±1.56) and the 1928 platform (1±1.59). This pattern was consistent across different-patient clustered pairs. However, no significant differences were observed among non-clustered isolate pairs [34].
Performance of the Decentralized CDST Pipeline
The CoDing Sequence Typer (CDST) presents a fully decentralized, MD5 hash-based framework that indexes predicted coding sequences (CDSs) and computes pairwise genomic distances without locus annotation or a central database. This approach eliminates the need for maintaining centralized reference databases while enhancing data privacy through irreversible MD5 hashing [65].
CDST was benchmarked against conventional typing methods using 1,961 complete Salmonella enterica genomes. The pipeline demonstrated high concordance with core-genome MLST (cgMLST), whole-genome MLST (wgMLST), core-genome SNP (cgSNP), Mash, and Split Kmer Analysis (SKA). The evaluation also identified three optimal clustering thresholds that correspond to different epidemiological and phylogenetic scales [65].
Table 2: Performance Benchmarks of CDST Versus Traditional Typing Methods
| Method | Concordance with CDST | Runtime Comparison | Storage Requirements | Key Applications |
|---|---|---|---|---|
| CDST | Reference | ~8× faster than cg/wgMLST | ~4% of original FASTA size | All levels of clustering |
| cgMLST | High | Baseline | ~25× original FASTA size | High-resolution typing |
| wgMLST | High | Slower than CDST | Higher than cgMLST | Comprehensive gene analysis |
| cgSNP | High | Variable by implementation | Moderate | High-sensitivity comparison |
| Mash | High | Fast | Minimal | Rapid large-scale comparisons |
| SKA | High | Intermediate | Minimal | Alignment-free analysis |
The CDST pipeline achieved approximately 8× faster runtimes than cg/wgMLST workflows and reduced storage requirements to approximately 4% of the original assembly FASTA size. Unsupervised clustering evaluation identified three optimal resolution levels: HC67 (outbreak-level), HC186 (lineage/serotype-level), and HC441 (global structure-level), which align well with conventional typing schemes. Cross-species validation on Listeria monocytogenes and Escherichia coli genomes confirmed that CDST recovers species-specific population structures without parameter adjustment [65].
Experimental Protocols and Methodologies
cgMLST Pipeline Comparison Protocol
The comparative study of cgMLST pipelines utilized 255 clinical isolates collected between 2018-2021 from patients at St. Jude Children's Research Hospital. Isolates were selected based on previous identification as being related to at least one other isolate using SeqSphere+ [34].
Wet-Lab Methodology:
- Isolate preparation, DNA extraction, and library preparation followed previously described protocols
- Illumina sequencing was performed to generate FASTQ files
- DNA extraction and library preparation were standardized across all samples
Bioinformatic Analysis:
- FASTQ files were uploaded and analyzed in parallel using three commercial platforms
- SeqSphere+: Utilized publicly available schemes from cgMLST.org for most species, with ad hoc schemes for Pseudomonas aeruginosa and Serratia marcescens
- 1928 Platform: Employed custom-developed allele-calling based on an alignment-free k-mer approach with cgMLST schemes created from NCBI RefSeq genomes
- ARESdb: Conducted de novo genome assembly, quality control, and cgMLST analysis with schemes from cgMLST.org
Statistical Analysis:
- Pairwise allelic differences were compared between pipelines
- Jonckheere-Terpstra tests evaluated allelic distance trends across categories
- Friedman tests and pairwise Wilcoxon signed-rank tests compared pipeline differences with Bonferroni correction
- All statistical tests were two-sided with a significance level of 0.05 [34]
CDST Validation Protocol
The validation of the CoDing Sequence Typer (CDST) pipeline employed 1,961 complete Salmonella enterica genomes from RefSeq to benchmark performance against conventional typing methods [65].
CDS Prediction and Hashing:
- Genome assemblies were annotated with Prodigal (v.2.6.3) to predict coding sequences
- Predicted CDSs were filtered to remove those containing ambiguous nucleotides and length <201 bp
- MD5 hashes were computed for each remaining CDS sequence
- Hash values were collected into lists for each sample and stored in a JSON database
Distance Calculation:
- Pairwise distances were estimated based on overlap of CDS hash lists
- Absolute distance (dijabs) = |Hi| - |Hi ∩ Hj|
- Relative distance (dijrel) = 1 - (|Hi ∩ Hj| / |Hi|)
- Symmetric distance matrix derived by taking minimum of directional relative distances
Comparative Benchmarking:
- wgMLST: Allele profiles generated using chewBBACA (v.3.3.9), processed with GrapeTree (v.1.5.0)
- cgMLST: chewBBACA profiles filtered to retain loci present in ≥95% of samples
- cgSNP: Genomes annotated with Prokka (v.1.14.6), core genome aligned with Roary (v.3.13.0), recombinant regions masked with Gubbins (v.2.4.1), SNP distances computed with snp-dists (v.0.8.2)
- Mash: SourMASH (v.4.8.14) with k=31 and scaling=1,000
- SKA: Default k-mer length of 15 [65]
Workflow Comparison: Centralized vs. Decentralized Typing
The following diagram illustrates the fundamental differences in workflow between traditional centralized cgMLST and the decentralized CDST approach:
Comparative Workflows: Centralized cgMLST vs. Decentralized CDST
Table 3: Key Research Reagent Solutions for Bacterial Typing Studies
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| Ridom SeqSphere+ | Commercial Software | cgMLST analysis using published schemes | High-resolution strain typing for outbreak investigation |
| 1928 Diagnostics | Commercial Platform | cgMLST with k-mer based allele calling | Automated pipeline for clinical pathogen analysis |
| ARES Genetics ARESdb | Commercial Platform | cgMLST with de novo assembly | Comprehensive resistance and virulence profiling |
| CDST Pipeline | Open-Source Tool | Decentralized hash-based typing | Privacy-preserving cross-laboratory surveillance |
| chewBBACA | Open-Source Tool | wgMLST allele calling | Whole-genome MLST schema development |
| Prodigal | Open-Source Tool | Coding sequence prediction | Essential first step in CDST and other gene-based methods |
| GrapeTree | Open-Source Tool | Phylogenetic tree visualization | Visualization of MLST and cgMLST results |
| SourMASH | Open-Source Tool | MinHash sketch comparisons | Rapid large-scale genome comparisons |
The benchmarking studies presented demonstrate that both commercial cgMLST pipelines and novel decentralized approaches like CDST provide effective solutions for bacterial subtyping, with performance characteristics that make them suitable for different research and public health contexts. The high concordance among cgMLST pipelines using suggested clustering thresholds supports their reliability for clinical outbreak investigation, despite differences in absolute allelic distances. Meanwhile, the decentralized CDST approach offers significant advantages in computational efficiency, data privacy, and interoperability across laboratories. These validation studies provide researchers with critical performance data to inform their selection of bacterial typing methodologies based on specific project requirements, whether for high-resolution outbreak investigation, broad population surveillance, or resource-constrained environments.
Molecular subtyping of bacterial pathogens has traditionally been the cornerstone of outbreak detection and investigation in public health microbiology. However, the application of these methods has expanded far beyond outbreak epidemiology to become fundamental tools for understanding population structure, evolutionary dynamics, and pathogen ecology. Two methodologies that have proven particularly valuable in these expanded applications are Multi-Locus Sequence Typing (MLST) and Comparative Genomic Fingerprinting (CGF). While both methods serve to differentiate bacterial strains, they operate on distinct principles and offer complementary insights into bacterial evolution and population biology. MLST targets the sequence variation in a carefully selected set of core housekeeping genes, providing a stable framework for understanding long-term evolutionary relationships and global population structure [35]. In contrast, CGF detects the presence or absence of accessory genomic elements, capturing more rapid evolutionary changes that may reflect adaptation to specific niches or environmental pressures [35] [5]. This comprehensive comparison examines the technical performance, methodological requirements, and research applications of CGF and MLST, providing researchers with the evidence base to select the most appropriate method for their specific investigations into bacterial population biology and evolution.
Fundamental Principles and Technical Comparison
Core Methodological Differences
The fundamental distinction between MLST and CGF lies in their genomic targets and the type of evolutionary information they capture. MLST schemes typically sequence approximately 450-500 base pair internal fragments of seven housekeeping genes that are essential for basic cellular functions and are thus universally present within a bacterial species [66]. The sequences of these fragments are compared to existing allele libraries in curated databases, and each isolate is assigned a sequence type (ST) based on its combination of alleles [67]. This approach provides a highly standardized and portable typing system that is ideal for global surveillance and long-term phylogenetic studies.
In contrast, CGF targets genes within the accessory genome – those genes not universally present in all strains of a species – which often include genes involved in environmental adaptation, virulence, and antimicrobial resistance [35] [5]. CGF typically employs multiplex PCR to detect the presence or absence of 40-83 carefully selected accessory genes, generating a binary fingerprint that can be used to cluster isolates based on shared accessory genome content [35] [5]. This approach captures a different type of genetic variation that may be more relevant for understanding short-term adaptation and functional differences between strains.
Table 1: Fundamental Characteristics of MLST and CGF
| Feature | Multi-Locus Sequence Typing (MLST) | Comparative Genomic Fingerprinting (CGF) |
|---|---|---|
| Genomic Target | Core housekeeping genes (7 loci) | Accessory genes (40-83 loci) |
| Type of Variation | Nucleotide sequence polymorphisms | Presence/absence of genes |
| Data Output | Allelic profile (sequence type) | Binary fingerprint (presence/absence profile) |
| Evolutionary Timescale | Long-term evolution | Short-term adaptation |
| Primary Applications | Population structure, phylogenetic analysis, long-term epidemiology | Outbreak detection, niche adaptation, functional gene analysis |
| Standardization | Highly standardized through international databases | Protocol-specific, though efforts toward standardization exist |
Resolution and Discriminatory Power
Studies directly comparing the resolution of MLST and CGF have demonstrated that CGF typically offers superior discriminatory power for distinguishing closely related isolates. Research on Campylobacter jejuni and C. coli found that while both methods provided good estimates of true phylogenetic relationships inferred from whole genome sequencing, CGF offered better differentiation of epidemiologically related isolates [35]. Similarly, in the development of a CGF assay for Arcobacter butzleri, the method demonstrated high Simpson's Index of Diversity values (>0.969), indicating exceptional ability to distinguish between strains [5].
The enhanced resolution of CGF stems from its targeting of the accessory genome, which generally evolves more rapidly than the core genome targeted by MLST. As noted by Taboada et al., "CGF is highly concordant with MLST, but with a better discriminatory power" [35]. This makes CGF particularly valuable for investigating suspected outbreaks where strains may be highly similar, or for differentiating endemic strains circulating in specific ecological niches.
Concordance with Genomic Phylogeny
When evaluated against whole genome sequencing as a gold standard, both MLST and CGF show strong concordance with genomic phylogeny, though they capture different aspects of evolutionary relationships. A comprehensive analysis of C. jejuni and C. coli genomes found that both MLST and CGF provided better estimates of true phylogeny than methods based on single loci, with adjusted Wallace coefficients demonstrating good agreement with a reference phylogeny based on highly conserved core genes [35].
The concordance between CGF and whole genome phylogenies can be remarkably high. In the development of a CGF40 assay for A. butzleri, researchers achieved an adjusted Wallace coefficient of 1.0 with respect to the reference phylogeny based on 72 accessory genes, indicating perfect agreement in cluster identification at the thresholds tested [5]. This demonstrates that carefully designed CGF assays can accurately reflect relationships inferred from more comprehensive genomic analyses.
Figure 1: Methodological Relationships and Primary Applications. MLST and CGF utilize different genomic regions (core vs. accessory) derived from whole genome sequencing, leading to complementary research applications with MLST excelling in population structure analysis and CGF offering advantages in outbreak detection.
Experimental Performance and Validation Data
Quantitative Performance Metrics
Direct comparisons of MLST and CGF across multiple bacterial species have yielded valuable quantitative data on their performance characteristics. A study evaluating subtyping methods for Campylobacter detection found that CGF appeared to be "one of the optimal methods for the detection of clusters of cases," though it could be beneficially supplemented by flaA SVR sequencing or MLST for certain applications [28]. The same study noted that different methods appeared to group isolates at different levels within the population, suggesting they might be optimal for different investigative purposes.
Table 2: Experimental Performance Comparison of MLST and CGF Based on Published Studies
| Performance Metric | MLST Performance | CGF Performance | Study Organism |
|---|---|---|---|
| Discriminatory Power | High (Simpson's ID ~0.90-0.95) | Very High (Simpson's ID >0.969) | Arcobacter butzleri [5] |
| Epidemiological Concordance | Good for long-term relationships | Excellent for recent outbreaks | Campylobacter jejuni [35] |
| Reproducibility | High (>99% for sequence-based analysis) | Very High (98.6% for presence/absence calls) | Arcobacter butzleri [5] |
| Concordance with WGS Phylogeny | Good (AWC 0.75-0.90) | Very Good to Excellent (AWC up to 1.0) | Campylobacter jejuni [35] |
| Typeability | High (>95% for quality sequences) | High (>95% for quality DNA) | Multiple species [35] [5] |
Concordance Analysis Between Methods
Studies have consistently demonstrated strong but incomplete concordance between MLST and CGF, reflecting their different genomic targets and evolutionary insights. Research on Campylobacter isolates found that CGF types showed "high concordance with MLST" while providing improved resolution [35]. This pattern of high concordance with enhanced resolution has been observed across multiple bacterial pathogens, making CGF particularly valuable for investigations requiring fine-scale differentiation of closely related isolates.
The relationship between CGF and MLST appears to follow consistent patterns. As noted by Taboada et al., "although MLST targets the sequence variability in core genes and CGF targets insertions/deletions of accessory genes, both methods are based on multi-locus analysis and provided better estimates of true phylogeny than methods based on single loci" [35]. This suggests that both methods benefit from the statistical robustness of sampling multiple independent genetic loci, despite targeting different types of variation.
Research Applications and Context-Specific Performance
Population Structure Analysis
MLST has established itself as the gold standard for investigating global population structure and long-term evolutionary relationships in bacterial pathogens. The method's stability, standardization, and extensive international databases make it ideal for classifying strains into clonal complexes and understanding their global distribution [8] [66]. For example, MLST analysis of Candida glabrata isolates from Kuwait identified 28 sequence types, including 12 novel STs, with ST46 being predominant across multiple hospitals [66]. This enabled researchers to track the geographic dissemination of successful clones and understand the population structure of this pathogen in a clinical setting.
CGF can complement MLST data in population studies by revealing fine-scale structure that may reflect recent ecological adaptations. While MLST excels at identifying broad phylogenetic relationships, CGF can detect subgroups within clonal complexes that may be associated with specific environmental niches, host adaptations, or virulence properties [5]. This makes CGF particularly valuable for understanding microevolution within successful lineages that may appear homogeneous by MLST.
Evolutionary Studies and Microevolution
CGF offers distinct advantages for studying recent evolutionary events and microevolution due to its targeting of the more dynamic accessory genome. The accessory genome often contains genes subject to strong selective pressures, such as those involved in antibiotic resistance, virulence, or environmental adaptation [35] [5]. By tracking changes in these genomic regions, CGF can reveal evolutionary adaptations occurring over shorter timescales than those captured by MLST.
MLST remains valuable for understanding broader evolutionary patterns and long-term phylogenetic relationships. The slower evolutionary rate of housekeeping genes makes MLST ideal for reconstructing the deep branching structure of bacterial phylogenies and classifying strains into evolutionarily meaningful groups [67] [66]. Studies of C. glabrata using MLST have demonstrated its utility in detecting "microevolution in hospital environment" and nosocomial transmission, highlighting its continued relevance for evolutionary studies [66].
Outbreak Detection and Investigation
While both methods have applications beyond outbreak detection, their performance characteristics make them differentially suited to epidemiological investigations. CGF's higher resolution makes it particularly valuable for detecting subtle relationships between isolates during outbreak investigations, where distinguishing transmission chains requires fine-scale differentiation [28]. The method's high throughput and relatively low cost compared to whole genome sequencing further enhance its utility for routine surveillance [35] [5].
MLST continues to play an important role in outbreak investigations by providing essential context for understanding how outbreak strains relate to the broader population structure of the pathogen [28]. The extensive curated databases available for many pathogens enable researchers to quickly determine whether an outbreak strain belongs to a recognized clonal complex with known epidemiological significance, such as the CG258 clonal group in Klebsiella pneumoniae [8].
Figure 2: Evolutionary Insights from Different Genomic Targets. The differential evolution rates of core housekeeping genes versus accessory genes provide complementary insights into bacterial evolution, with MLST capturing stable phylogenetic signals and CGF revealing adaptive changes.
Methodological Protocols and Technical Considerations
Standard MLST Workflow
The MLST protocol follows a standardized approach across bacterial species, though specific gene targets and amplification conditions are tailored to each pathogen. The general workflow includes:
DNA Extraction: High-quality genomic DNA is extracted from pure bacterial cultures using commercial kits or standardized protocols [28] [66]. DNA quality and concentration are verified using spectrophotometry or fluorometry.
PCR Amplification: Approximately 450-500 bp fragments of seven housekeeping genes are amplified using pathogen-specific primers [66]. Reaction conditions are optimized for each gene target, typically involving 25-35 amplification cycles with gene-specific annealing temperatures.
DNA Sequencing: PCR products are purified and sequenced in both directions using the same primers as for amplification [66]. Sanger sequencing remains the gold standard, though next-generation sequencing platforms are increasingly employed.
Sequence Analysis and Allele Assignment: Sequences are trimmed and assembled, then compared to curated databases such as PubMLST (http://pubmlst.org) for allele assignment [28] [66]. Contiguous sequences for each locus are compared to existing alleles, and new alleles are submitted for verification and curation.
Sequence Type Determination: The combination of alleles across the seven loci defines the sequence type (ST). Novel combinations are assigned new ST numbers following database submission and verification [66].
CGF Methodology Development and Implementation
CGF assay development involves a more complex initial phase of target selection and validation, followed by a streamlined typing protocol:
Target Selection: Comparative genomic analysis of diverse strains identifies accessory genes with variable presence across the population [35] [5]. Bioinformatic tools like CGF Optimizer select optimal gene sets that maximize discrimination and concordance with reference phylogenies.
Primer Design and Validation: Multiplex PCR primers are designed for each target gene and validated for specificity and amplification efficiency [5]. Primer sets are combined into multiplex panels, typically targeting 40-60 genes across several reactions.
CGF Profiling: Genomic DNA is amplified using the multiplex PCR panels, and amplification products are detected using capillary electrophoresis or microarray platforms [35] [5]. The presence or absence of each target gene is recorded as a binary score.
Data Analysis and Cluster Identification: Binary profiles are analyzed using similarity coefficients (e.g., Jaccard) and clustered using methods such as UPGMA or neighbor-joining [5]. Isolates are grouped into clades based on profile similarity, with thresholds typically set at 90-95% similarity.
Profile Database Management: CGF profiles are stored in specialized databases that allow for comparison across studies and laboratories [5]. Standardized nomenclature facilitates data exchange, though universal databases are less established than for MLST.
Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for MLST and CGF
| Reagent/Platform | Function | MLST Application | CGF Application |
|---|---|---|---|
| Commercial DNA Extraction Kits | High-quality genomic DNA isolation | Required for PCR amplification | Required for multiplex PCR |
| Pathogen-Specific Primers | Target gene amplification | 7 pairs for housekeeping genes | 40-83 pairs for accessory genes |
| PCR Reagents | Amplification of target sequences | Standard and gradient PCR | Multiplex PCR optimization |
| DNA Sequencing Platform | Sequence determination | Sanger or NGS platforms | Generally not required |
| Capillary Electrophoresis | Separation of amplification products | For verification only | Essential for fragment detection |
| Curated Database | Strain comparison and classification | PubMLST and species-specific databases | Laboratory-specific with standardization efforts |
| Bioinformatics Software | Data analysis and phylogenetics | eBURST, Phyloviz, BioNumerics | Custom scripts, CGF Optimizer, BioNumerics |
Discussion: Integration with Whole Genome Sequencing and Future Directions
The rapid advancement of whole genome sequencing technologies is transforming the landscape of bacterial subtyping, with both MLST and CGF finding new roles within the genomic era. Core genome MLST (cgMLST) and whole genome MLST (wgMLST) approaches are extending the MLST concept to encompass hundreds or thousands of genes across the core and accessory genome [8] [63]. Similarly, CGF principles are being incorporated into gene content analysis pipelines that examine the entire accessory genome rather than predefined gene sets [35] [5].
Studies comparing these methods with whole genome sequencing have demonstrated that both MLST and CGF show strong concordance with genomic phylogenies, while each capturing different aspects of evolutionary relationships [35] [8]. As noted in one evaluation, "cgMLST and coreSNP are more discriminant than PFGE, and both approaches are suitable for transmission analyses" [8], suggesting that next-generation methods building on both MLST and CGF concepts will dominate future subtyping applications.
The choice between MLST and CGF—or decisions about incorporating newer genomic methods—should be guided by specific research questions, available resources, and the need for comparability with existing data. MLST remains the preferred method for global surveillance, population genetics, and studies requiring extensive database comparisons [67] [66]. CGF offers advantages for high-resolution outbreak investigation, niche adaptation studies, and research focused on accessory genome dynamics [35] [5]. As sequencing costs continue to decline, both methods will likely be increasingly applied as in silico analyses from whole genome sequence data rather than as standalone laboratory protocols [35] [68].
MLST and CGF represent complementary approaches to bacterial subtyping that offer distinct insights into population structure and evolutionary biology. MLST provides a stable, standardized framework for understanding broad phylogenetic relationships and classifying strains into evolutionarily meaningful groups, making it ideal for global surveillance and population genetics studies. CGF offers higher resolution and sensitivity for detecting recent evolutionary changes and adaptations, particularly those involving the accessory genome, making it valuable for outbreak investigation and studies of microevolution.
The expanding applications of both methods beyond outbreak detection to fundamental questions in bacterial evolution and ecology underscore their continued relevance in the genomic era. Rather than viewing them as competing technologies, researchers should recognize their complementary strengths and select the method—or combination of methods—that best addresses their specific research questions. As bacterial subtyping continues to evolve, the principles embodied in both MLST and CGF will undoubtedly inform the next generation of genomic analysis methods that further advance our understanding of bacterial population biology and evolution.
Conclusion
The comparative analysis of CGF and MLST reveals that CGF often provides superior discriminatory power for detecting epidemiologically relevant clusters, making it highly suitable for outbreak investigations and real-time surveillance. However, MLST remains invaluable for long-term population structure studies and maintaining universal nomenclature. The choice between methods is not a question of which is universally better, but which is fit-for-purpose. The future of bacterial subtyping lies in the integration of these methods with whole-genome sequencing, leveraging core-genome MLST (cgMLST) and coreSNP analysis for unprecedented resolution. As public health laboratories worldwide transition to genomic surveillance, a combined approach that utilizes the speed of CGF and the context of MLST within a WGS framework will be crucial for effectively tracking and controlling the spread of bacterial pathogens.
References
- 1. Multi-Locus Sequence Typing [https://kb.ezbiocloud.net/home/science-blogs/identify/multi-locus-sequence-typing]
- 2. MLST — OH SFS Handbook v0.0.1 documentation [https://oh-sfs-handbook.readthedocs.io/en/latest/Typing/MLST.html]
- 3. Multi-Locus Sequence Typing [https://www.cd-genomics.com/microbioseq/multi-locus-sequence-typing.html]
- 4. Development and Validation of a Comparative Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3295178/]
- 5. Development of a comparative genomic fingerprinting assay ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-015-0426-4]
- 6. A Framework for Assessing the Concordance of Molecular ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2012.00057/full]
- 7. Multilocus Sequence Analysis of Housekeeping Genes and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3766922/]
- 8. Comparison of core-genome MLST, coreSNP and PFGE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7276701/]
- 9. Comparison of a semi-automated rep-PCR system and ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701210000151]
- 10. Rapid and Routine Molecular Typing Using Multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9002326/]
- 11. Assessment and Comparison of Molecular Subtyping ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.01591/full]
- 12. Comparative analysis of core genome MLST and SNP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5899760/]
- 13. Unraveling the impact of genome assembly on bacterial typing [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10982-z]
- 14. In silico and in vitro comparative analysis of 79 Acinetobacter ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12210950/]
- 15. Metagenomic Strain-Typing Combined with Isolate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9860660/]
- 16. A new approach for establishing multilocus sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513078/]
- 17. Validation of Core and Whole-Genome Multi-Locus ... [https://www.mdpi.com/2076-2607/13/6/1310]
- 18. Genomic analysis of the main epidemiological lineages ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2024.1499839/full]
- 19. Genome MLST scheme for tracing genetic diversity and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12305728/]
- 20. MentaLiST – A fast MLST caller for large MLST schemes [https://pmc.ncbi.nlm.nih.gov/articles/PMC5857373/]
- 21. MiST: rapid, accurate and flexible (core-genome) multi-locus ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12324-z]
- 22. of classical multi-locus sequence typing software for... Comparison [https://pmc.ncbi.nlm.nih.gov/articles/PMC5610716/]
- 23. accurate and ultrafast genomic profiling with exact sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7430640/]
- 24. GitHub - katholt/ srst : Short Read Sequence Typing for 2 ... Bacterial [https://github.com/katholt/srst2]
- 25. : Rapid genomic surveillance for public health and hospital... SRST 2 [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-014-0090-6]
- 26. Comparisons between SRST2, ARIBA, and KmerResistance [https://www.microbialsystems.cn/en/post/comparing_ariba_srst2_kmerresistance/]
- 27. STRAIN: an R package for multi-locus sequence typing from whole... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2887-1]
- 28. Comparison of Molecular Typing Methods Useful for Detecting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3295161/]
- 29. Techniques in bacterial strain typing: past, present, and future [https://pmc.ncbi.nlm.nih.gov/articles/PMC9245535/]
- 30. Multi-Locus Sequence Typing (MLST) [https://docs.omicsbox.biobam.com/latest/Multi-Locus-Sequence-Typing/]
- 31. kakapo: easy extraction and annotation of genes from raw ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10688300/]
- 32. Tensile Strength, Cellular Content and Degradation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12044792/]
- 33. CHROMATOGATE: A TOOL FOR DETECTING BASE MIS ... [https://www.sciencedirect.com/science/article/pii/S2001037014601100]
- 34. Comparison of core genome multi-locus sequencing typing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506026/]
- 35. PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3417556/]
- 36. Inquiring into the Gaps of Campylobacter Surveillance ... [https://www.mdpi.com/2306-7381/4/3/36]
- 37. Comparative genomic fingerprinting of Campylobacter [https://pmc.ncbi.nlm.nih.gov/articles/PMC9507621/]
- 38. Development and validation of a comparative genomic ... [https://pubmed.ncbi.nlm.nih.gov/22170908/]
- 39. High-Resolution Melting System to Perform Multilocus ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0016167]
- 40. Source attribution [https://en.wikipedia.org/wiki/Source_attribution]
- 41. Typing methods based on whole genome sequencing data [https://pmc.ncbi.nlm.nih.gov/articles/PMC7993478/]
- 42. Development of a comparative genomic fingerprinting assay for rapid... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4424573/]
- 43. a modified MLST typing tool based on short read sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC4991843/]
- 44. Low coverage whole genome sequencing enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6880438/]
- 45. The hidden genetic reservoir: structural variants as drivers of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12379486/]
- 46. A comparison of methods for detecting DNA methylation from ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03207-9]
- 47. Real-World Performance of a Comprehensive Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8384401/]
- 48. Galaxy @Sciensano: a comprehensive bioinformatics portal ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-11182-5]
- 49. Strain-level characterization of bacterial pathogens using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12542171/]
- 50. What's Your Subtype? The Epidemiologic Utility of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5697907/]
- 51. Comparison of Three Molecular Methods Used for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1248497/]
- 52. Assessment and Comparison of Molecular Subtyping and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6639432/]
- 53. Microbial Sequence Typing in the Genomic Era - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5908768/]
- 54. Multilocus Sequence Typing Compared to Pulsed-Field Gel Electrophoresis for Molecular Typing of Pseudomonas aeruginosa - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2168506/]
- 55. Comparative Analysis of Multilocus Sequence Typing and Pulsed-Field Gel Electrophoresis for Characterizing Listeria monocytogenes Strains Isolated from Environmental and Clinical Sources - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC321703/]
- 56. Comparative Analysis of Subtyping Methods against a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4290925/]
- 57. A Practical Bioinformatics Workflow for Routine Analysis of ... [https://www.mdpi.com/2076-2607/10/12/2364]
- 58. Comparative analysis of IR-Biotyper, MLST, cgMLST ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10986520/]
- 59. Comparing core-genome MLST with PFGE and ... [https://www.sciencedirect.com/science/article/pii/S2213716522001503]
- 60. Comparative analysis of core genome MLST and SNP ... [https://www.sciencedirect.com/science/article/pii/S0168160518300746]
- 61. Comparison of Whole Genome (wg-) and Core ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.01729/full]
- 62. Comparison of SNP-based subtyping workflows for bacterial ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0192504]
- 63. Detailed Evaluation of Data Analysis Tools for Subtyping ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.02897/full]
- 64. Development of a novel core genome MLST scheme for ... [https://www.nature.com/articles/s41467-022-31908-x]
- 65. CoDing Sequence Typer (CDST): a fast, simple, decentralized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548756/]
- 66. Molecular fingerprinting by multi-locus sequence typing ... [https://www.frontiersin.org/journals/public-health/articles/10.3389/fpubh.2023.1242622/full]
- 67. Bacterial typing methods from past to present [https://www.sciencedirect.com/science/article/abs/pii/S2452014422001832]
- 68. Genome-wide approaches to bacterial strain typing [https://pmc.ncbi.nlm.nih.gov/articles/PMC12237114/]